بازنگری لوسمیهای خانواده مایلوئیدی (WHO 2016)
قسمت سوم
دكتر حبیبالله گلافشان عضو هيئت علمي دانشگاه علوم پزشكي شيراز
نگين شكرگزار كارشناس ارشد هماتولوژی و بانک خون دانشگاه علوم پزشكي شيراز
سندرم همپوش مایلوپرولیفراتیو/ مایلودیسپلاستیک با سیدروبلاست حلقوی و ترومبوسیتوز
(MDS/MPN-RS-T)
سندرم همپوش فوق قبلاً تحت عنوان کمخونی رفراکتوری با سیدروبلاست حلقوی و ترومبوسیتوز نامگذاری شده بود. حضور 15 درصد یا بیشتر سیدروبلاست حلقوی در مغز استخوان برای تشخیص ضروری است. در سیدروبلاست حلقوی گرانولهای آهن حداقل یکسوم محیط هسته را مانند یقه پوشش دادهاند. جهش SF3B1 که با سیدروبلاست حلقوی در ارتباط است، با حضور تغییرات دیسپلاستیک تشخیص را قطعی میکند. جهش فوق اغلب همراهی با جهش Jak2V617F داشته و در کمتر از 10% موارد با جهشهای CARL یا MPL همراه است.
معیارهای تشخیصی MDS/MPN-RS-T
1- کمخونی به همراه ترومبوسیتوز و دیسپلازی رده اریتروئیدی با یا بدون دیسپلازی چندردهای، ≥15% سیدروبلاست حلقوی، کمتر از یک درصد بلاست در خون محیطی و کمتر از 5 درصد در مغز استخوان
2- تداوم افزایش شمارش پلاکت ≥450×109/L
3- حضور جهش SF3B1 و در نبود جهش بایستی بیمار سابقه تجویز داروی سایتوتوکسیک یا فاکتور رشد نداشته باشد.
4- منفی بودن BCR/ABL، نبود بازآرایی ژنهای فاکتور رشد پلاکتی A و B و منفی بودن PCM1/Jak2، t(3;3) یا inv(3) یا del(5q)
نئوپلاسمهای مایلوئیدی با اختلالات کروموزومی سلولهای زایا (germ line)
با وجود اینکه بیشتر موارد لوسمی AML و MDS پراکندگی دارند، ولی زیرگروههایی از لوسمیها همراه با جهشهای سلولهای زایا و خانوادگی هستند. طبقهبندی WHO در سال 2016، عنوان لوسمیها و سندرمهای مایلودیسپلاستیک در زمینه جهشهای رده زایا را به طبقهبندی اضافه کرده است. جهشهای germ line تنها به بیمار مبتلا ختم نمیشود، بلکه بایستی به اعضای خانواده آگاهی داد و آنها را برای جهشهای خاص اسکرین کرد.
طبقهبندی نئوپلاسمهای مایلوئیدی در زمینه جهشهای سلولهای زایا (germ line)
- نئوپلاسمهای مایلوئیدی در زمینه جهشهای germ line بدون اختلال قبلی یا نقص ارگان مانند لوسمی AML با جهش germ line در فاکتور نسخــــــهبرداری CEBPA و یا نئوپلاسمهای مایلــــــــــوئیدی با جهش DDX41 (ATP dependent RNA helicase)
- نئوپلاسمهای مایلوئیدی با جهش germ line در زمینه اختلال پلاکتی مانند نئوپلاسمهای مایلوئیدی با جهش در فاکتور نسخهبرداری Runx1، نئوپلاسمهای مایلوئیدی با جهش در ANKDR26 و نئوپلاسمهای مایلوئیدی با جهش ETV-6
- نئوپلاسمهای مایلوئیدی در زمینه جهشهای germ line و اختلال ارگان مانند نئوپلاسمهای مایلوئیدی با جهش در GATA2، نئوپلاسمهای مایلوئیدی با سندرمهای ناکارایی مغز استخوان (bone marrow failure)، نئوپلاسمهای مایلوئیدی با اختلالات بیولوژی تلومر، لوسمی مزمن مایلومونوسیتیک جوانی (JMML) همراه با نوروفیبروماتوز و سندرم نونان و نئوپلاسمهای مایلوئیدی همراه با سندرم داون
از مهمترین جهشهای ژنی در سندرم نونان میتوان به PTPN11، SOS1، RAF1 و KRAS اشاره کرد. نوروفیبروماتوز گروهی از اختلالات ژنتیکی هستند که تومور روی سیستم عصبی رشد میکند.
سندرم داون 10 تا 20 برابر شانس ابتلا به ALL در سه دهه اول زندگی را افزایش میدهد و در زیر یک سالگی، شانس ابتلا به AML زیاد است. لوسمیهای ALL با پیشآگهی مطلوب مانند هایپردیپلوئیدی و فیوژن ETV6/Runx1 در 10 تا 20% موارد در مقایسه با 50% از بیماران بدون سندرم داون رخ میدهد. در لوسمی لنفوبلاستیک مبتلایان به سندرم داون فعالیت ژنهای CRLF2 (cytokine receptor life factor2)، Jak2 و حذف IZKF1 شایع است. فاکتور CRLF2 قادر به فعالسازی مسیر علائمرسانی از طریق Jak2 و Stat است و بازآرایی آن با ژن زنجیره سنگین ایمونوگلوبولین روی کروموزوم 14 یا P2Y (ژن پورینرژیک) موجب لوسمی لنفوبلاستیک نوع B در سندرم داون میگردد. فرآورده ژن CRLF2، گیرنده لنفوپویتین استرومال تیموس (TSLP) است و جایگاه ژن کروموزوم X است.
کمتر از 5% لوسمی حاد لنفوبلاستیک در بچهها در زمینه وراثت اختلال ژنتیکی است. جهش germ line در TP53 (ژن سرکوبکننده تومور) که موجب سندرم Li-fraumeni میشود، خانواده را مستعد انواع تومورها میکند. جهش TP53 در ALL با بلاستهای هایپودیپلوئیدی (32-39 کروموزوم) همراه است.
گفتنی است که t(12;21) در 1% نمونه بند ناف نوزادان نرمال مثبت است، اما تعداد بسیار کمی در سالهای بعد مبتلا به ALL میگردند چون سرطانی شـدن سلول، چندمرحلهای است. در این جابهجایی، ژنهای ETV-6 و CBFα یا Runx1 در یکدیگر ادغام میگردند. ژن ETV-6 (ets variant 6) یا ژن TEL، رمزدهنده فــــــــــــــاکتور نسخهبرداری از خانواده ETS (E twenty six) است.
لوسمیهای حاد مایلوئیدی (Acute Myeloid Leukemia)
بازنگری طبقهبندی AML در سال 2016 همان طبقهبندی 2008 با تغییرات اندک مورد قبول قرار گرفته است. در جدول زیر، طبقهبندی جدید 2016 لوسمیهای حاد مایلوبلاستیک به شرح زیر است:

تغییر نام ژن MLL به KMT2A و پیشآگهی مطلوب AML ناشی از جهش فاکتور نسخهبرداری CEBPA در هر دو آلل و جای دادن AML با جهش NPM1 (نوکلئوفسمین) با پیشآگهی مطلوب در جدول مشاهده میشود.
نکته قابل توجه اینکه وارونگی inv(3) یا جابهجایی t(3;3)، بیانگر ادغام ژن نیست بلکه با جابهجا کردن GATA2 موجب تشدید بیان ژن MECOM میشود. ژن MECOM(MDS and EV11 Complex)، پروتئین نسخهبرداری در هسته سلول را رمزدهی میکند که نقش مهمی در مسیر علائمرسانی دارد و بیان ناهنجار آن با لوسمی AML، سندرم مایلودیسپلاستیک و لوسمی مزمن مایلوسیتیک با پیشآگهی نامطلوب همراه است.
لوسمی حاد مایلوبلاستیک با تغییرات دیسپلاستیک
گفتنی است که حضور تغییرات دیسپلاستیک (multilineage) در لوسمی حاد مایلوبلاستیک در مواردی که با جهشهای NPM1 یا جهش هر دو آلل CEBPA همراه باشد، آن را در گروه لوسمی حاد مایلوئیدی با دیسپلازی قرار نمیدهد بلکه در نبود جهشهای فوق و حضور سلولهای دیسپلاستیک 50% یا بیشتر، در حداقل دو رده سلولی میتوان تشخیص AML با تغییرات دیسپلازی با پیشآگهی نامطلوب را داد.
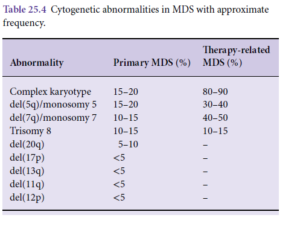
حضور تاریخچه MDS و اختلال ژنتیکی مربوط به سندرم مایلودیسپلاستیک بهجز حذف 9q کافی است که لوسمی حاد مایلوبلاستیک با دیسپلازی تعریف گردد. گفتنی است که حذف 9q با حذف NPM1 و جهش دو آلل CEBPA همراهی دارد. در جدول زیر اختلالات سیتوژنتیک که AML را با تغییرات دیسپلاستیک تعریف میکند را مشاهده میکنید.
لوسمیهای حاد مایلوبلاستیک که در قالب کلاسهبندی اختلالات ژنتیکی نمیگنجد (AML-NOS)
در بازنگری 2016 مانند کلاسهبندی 2008 سازمان بهداشت جهانی(WHO)، چنانچه لوسمی حاد مایلوبلاستیک دارای اختلالات کروموزومی تکرارشونده نباشد، بنا به مرفولوژی FAB (French American British) طبقهبندی میگردد؛ با این تفاوت که لوسمی حاد M6(Erythroid/Myeloid) از کلاسهبندی حذف گردیده و دارای بازنگری به شرح زیر است:
لوسمی AML-M6 در کلاسهبندی 2008 برمبنای حضور ≥50% از پیشسازهای اریتروئیدی و ≥20% میلوبلاست در میان جمعیت غیراریتروئیدی سلولهای مغز استخوان بوده است. در طبقهبندی جدید، درصد سلولهای میلوبلاست بهعنوان درصدی از کل سلولهای هستهدار مغز استخوان شمرده میشود و ازاینرو دارای تغییرات نامگذاری به شرح زیر است:
| پیشسازهای اریتروئیدی | درصد میلوبلاست از کل سلولها | اختلال ژنتیکی تکرارشونده | نامگذاری جدید |
| ≥50% | ≥20% | بله | لوسمی AML با اختلال کروموزومی |
| ≥50% | ≥20% | خیر | AML-MRC (چنانچه با تغییرات دیسپلاستیک همراه باشد) |
| ≥50% | ≥20% | خیر | AML-NOS (بدون تغییرات دیسپلاستیک) |
| ≥50% | <20% اما ≥20% از سلولهای غیراریتروئیدی | خیر |
MDS با افزایش بلاست
|
| ≥80% | <20% | خیر |
AML (pure erythroid) |
MRC = Myelodysplasia related changes
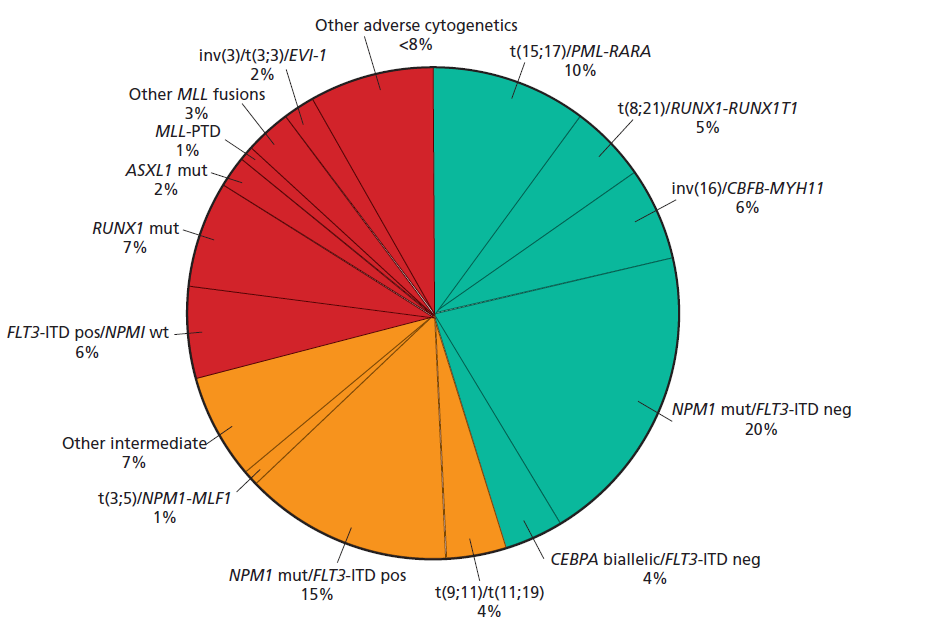
در نمودار دایرهای فوق درصد اختلالات کروموزومی در لوسمیهای حاد مایلوئیدی مشاهده میگردد
سارکوم مایلوئیدی (myeloid sarcoma)
سارکوم مایلوئیدی بهعنوان یک تظاهر ویژه از هر زیرگروه از AML در طبقهبندی جدید مانند طبقهبندی سال 2008 نگه داشته شده است. به تجمع غده مانند سلولهای بلاست مایلوئیدی در زیر پوست یا در هر بافت دیگر سارکوم مایلوئیدی گویند.
سارکوم مایلوئیدی ممکن است از ابتدا همراه با سلولهای لوسمی در خون محیطی و مغز استخوان یا بهعنوان نشانه عود لوسمی مایلوبلاستیک یا نشانه پیشرفت قبلی سندرمهای مایلودیسپلاستیک و یا تبدیل نئوپلاسمهای مایلوپرولیفراتیو به لوسمی حاد مایلوبلاستیک باشد. مواردی از سارکوم مایلوئیدی که در آن شواهدی از درگیری مغز استخوان نیست، بایستی کاملاً ارزیابی گردد و ازاینرو میتوان آن را در زیرگروه مخصوص AML قرار داد. آسپیره از سارکوم مایلوئیدی یا اسلاید رنگشده از لمس سلولهای غده (touch prep)، تجمع سلولهای بلاست را با ویژگی مایلوبلاست یا مونوبلاست نشان میدهد.
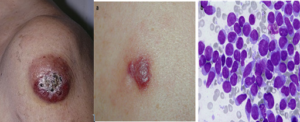
سارکوم مایلوئیدی از سلولهای بلاست مایلوئیدی یا مونوسیتی بهصورت غده مانند در زیرپوست یا هر بافت نرم دیگر ممکن است مشاهده گردد. نمای میکروسکوپی انبوهی از سلولهای بلاست با خاصیت پروکسیداز مثبت را نشان میدهد. سارکوم مایلوئیدی ممکن است بهصورت اولیه یا در همراهی با سایر لوسمیهای مایلوئیدی ظاهر گردد و مشاهده آن علامت عود یا تبدیل به مرحله حاد لوسمی است
تکثیر سلولهای مایلوئیدی در سندرم داون
تکثیر سلولهای مایلوئیدی در سندرم داون شامل مایلوپرولیفراتیو موقتی (TAM) و لوسمی حاد مایلوئیدی است. هر دو مورد معمولاً در خانواده لوسمی مگاکاریوبلاستیک بوده و سندرم TAM در بدو تولد یا چند روز پس از تولد رخ داده و در طول یک تا دو ماه فروکش میکند. لوسمی حاد مایلوئیدی در سه سال اول زندگی با سابقه قبلی TAM یا بدون آن در اکثر بیماران با سندرم داون رخ داده و چنانچه درمان نشود، سیری مانند لوسمی در بزرگسالان دارد. سندرمهای TAM و لوسمی حاد در سندرم داون با جهش GATA1 و جهش مسیر علائمرسانی Jak/Stat و اختلالات دیگر کروموزومی همراه است.
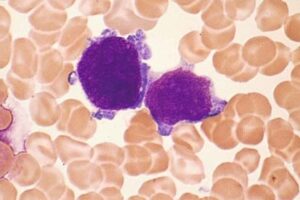
سلولهای بلاست با جوانههای سیتوپلاسمی با احتمال مگاکاریوبلاست در سندرم مایلوپرولیفراتیو موقتی در سندرم داون
لوسمیهای حاد با رده مبهم سلولی (ambiguous lineage)
گرچه دادههای بالینی و آزمایشگاهی در مورد این دسته از لوسمیها ابتدایی است، ولی به نظر میرسد که لوسمیهای با فنوتایپ مخلوط (mixed) MPAL با جابهجایی t(9;22) پاسخ بهتری به بازدارندههای تیروزین کیناز میدهند.
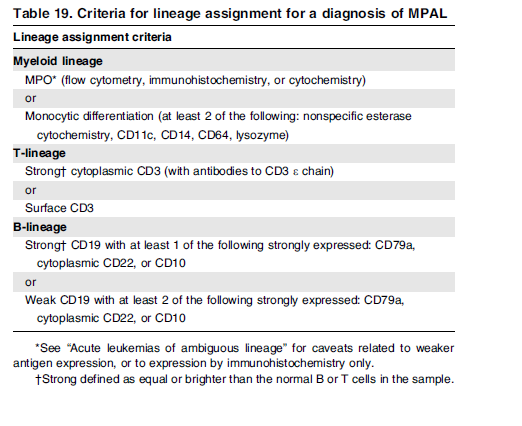
هرگونه ترکیب لوسمی حاد (Mixed acute leukemia) امکانپذیر است، مانند لوسمی B/myeloid، T/myeloid و حتی بهندرت B/T/myeloid که در این میان گزارشها از شیوع بیشتر B/myeloid حکایت دارد. توجه داشته باشید که برای تشخیص این گونه لوسمیها نیاز به پانل گسترده آنتیبادیهای مونوکلونال با فلوسیتومتر چند پارامتری و کاربرد روشهای ایمونوهیستوشیمی است. گاهی لنفوبلاست B بیان ضعیفی از میلوپروکسیداز را نشان میدهد که این بیان ضعیف، لوسمی را در گروه لوسمی مخلوط حاد قرار نمیدهد. جابهجاییهای t(9;22) و MLL 11q23 با شیوع بیشتری با لوسمیهای میکسد همراهی دارند. جدول زیر مارکرهای ردههای اختصاصی برای تشخیص لوسمیهای با فنوتایپ میکسد را نشان میدهد.
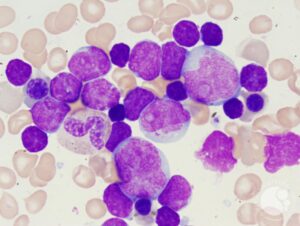

لوسمی با سلولهای میکسد (Mixed) با نمای لنفوبلاستیک و مایلوبلاستیک مشاهده میگردد.
آنالیز ایمونوفنوتایپ با فلوسایتومتری حضور هر دو نوع بلاست را نشان میدهد
لوسمیهای حاد لنفوبلاستیک
بازنگری طبقهبندی لوسمیهای حاد لنفوبلاستیک به علت اختلالات ژنتیکی در تابلوی زیر مشاهده میشود:

لوسمی لنفوبلاستیک با بلاستهای هایپودیپلوئید، اغلب در همراهی با جهش P53 است. گفتنی است که لنفوبلاستهای هایپردیپلوئیدی (51 تا 65 کروموزوم) شایعترین اختلال کروموزومی در لوسمی است که در حدود 35% لوسمی بچهها و حدود 10% لوسمی بزرگسالان مشاهده میشود. این اختلال ژنتیکی به علت حساس بودن سلولهای سرطانی به متوتروکسات دارای پیشآگهی مطلوب است، درحالیکه برخی معتقد به تریزومیهای 4،10 و 17 بوده و برخی خطر عود کم را در رابطه با تریزومی 18 در این حالت مطرح کردهاند. برخلاف پاسخ مطلوب اختلال فوق، چنانچه تعداد کروموزومهای سلولهای بلاست نزدیک تریپلوئیدی باشد (69-81)، پاسخ به درمان شبیه موارد غیرهایپردیپلوئیدی است. در مواردی که تعداد کروموزومها نزدیک تتراپلوئیدی باشد (82-94)، به احتمال زیاد ایمونوفنوتایپ T دارند. بلاستهای هایپودیپلوئید با کمتر از 45 کروموزوم در کمتر از 2% موارد ALL با پیشآگهی نامطلوب رخ میدهد، بهویژه آن دسته از بیماران که تعداد کروموزومها کمتر از 30 (نزدیک هاپلوئید) یا بین 30 تا 39 (near hypodiploid) باشد. تعداد کروموزومها با اندکس DNA با رنگآمیزی فلورسانس یا آنتیبادی مونوکلونال علیه سانترومر قابل شناسایی است.
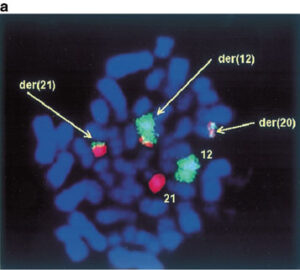
لوسمی حاد لنفوبلاستیک با جابهجایی t(12; 21) در غالب موارد فنوتایپ early pre B و هایپردیپلوئید پیشآگهی مطلوب دارد
لوسمی لنفوبلاستیک لنفوسیتهای B با تکثیر داخل کروموزومی 21 (iAMP21)
این لوسمی با تکثیر قطعهای از کروموزوم 21 با پروب برای ژن Runx1 توسط روش FISH قابل شناسایی است که 5 یا بیشتر کپی RUNX1 روی یک کروموزوم 21 در مرحله متافاز را نشان میدهد. این اختلال ژنتیکی در حدود 2% بچههای مبتلا به ALL بهویژه بچههای بزرگتر با شمارش WBC کم رخ میدهد. این لوسمی تهاجمی بوده و پیشآگهی نامطلوب دارد و ازاینرو نیاز به درمان تهاجمی دارد.
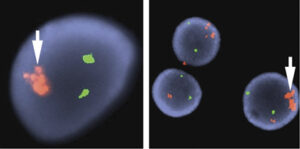
تکثیر چندتایی داخل کروموزومی ژن RUNX1 در مواردی از لوسمی حاد لنفوبلاستیک مشاهده شده است که با پروب RUNX1 میتوان آن را بهصورت ناحیه توسعه دادهشده داخل کروموزومی مشاهده کرد
لوسمی لنفوبلاستیک از خانواده لنفوسیتهای B با جابهجایی ژنهای تیروزین کیناز یا گیرندههای سایتوکاین (BCR/ABL1-like ALL)
اهمیت این گروه از لوسمیهای ALL، تهاجمی بودن و با این حال امکان پاسخ به داروهای ضد تیروزین کیناز (Tki) است. از ویژگیهای BCR/ABL1-like ALL جابهجاییهای ژنهای دیگر تیروزین کیناز یا جابهجایی CRLF2 (cytokine receptor like factor2) است. جابهجایی CRLF2 در اغلب موارد همراه با جهش Jak2 بوده و بهویژه در لوسمی ALL مبتلایان به سندرم داون مشاهده میگردد. این جابهجایی موجب میشود که فرآورده پروتئینی ژن CRLF2 با افزایش بیان گیرنده لنفوپویتین سلولهای استرومال تیموس (TSLPR) همراه گردد که با روش فلوسیتومتری قابل تشخیص است.
ژنهای تیروزین کیناز شامل ABL2، PDGFRB، NTRK3 (neurotropic receptor tyrosin kinase)، TYK2، CSF1R (گیرنده رشد کلنی) و Jak2 میباشند که با ژنهای متعدد دیگری قابلیت ادغام دارند.
ادغام EBF1-PDGFRB (early B cell factor) در بیماران ALL پاسخ مناسبی به بازدارندههای تیروزین کیناز میدهد. بیماران مبتلا به ALL با اختلالات کروموزومی شبیه BCR/ABL1 با شیوع فراوان کارایی ژنهای IKZF1 و CDKN2A/B را از دست دادهاند. ژن IKZF1 (Ikaros zinc finger 1) نقش مهمی در هماتوپویز و سرکوب B-ALL دارد و از دست دادن آن شخص را مستعد به ALL میکند. یک پلیمرفیسم (SNP) در ناحیه 3ʹ ژن، علاوه بر ALL با دیابت تایپ یک همراه است.
ژن CDKN2 (cycline dependent kinase inhibitor) روی کروموزوم 9 بوده و دو فرآورده پروتئینی P16 (از خانواده INK4) و P14arf دارد که هر دو سرکوبکننده تومور از طریق فعال کردن ژنهای رتینوبلاستوما (Rb) و P53 میباشند.
لوسمی لنفوبلاستیک خانواده T (T-ALL)
حدود 15% بچهها و بزرگسالان مبتلا به ALL دارای فنوتایپ T میباشند. شیوع فنوتایپ T با افزایش سن بیشتر میشود. سلولهای T مارکرهای CD7 و CD3 (اغلب سیتوپلاسمی) را بیان میکنند. لوسمی حاد لنفوبلاستیک بر اساس ایمونوفنوتایپ به سه دسته اولیه (early) با مارکرهای –CD7+، CD3+، CD4 و –CD8 و حدواسط (mid or common) با مارکرهای +CD3، و +CD4+، CD8 و +CD1a و نهایی (late) با مارکرهای CD3 سطحی و یکی از مارکرهای CD4 یا CD8 طبقهبندی شده است. گیرنده TCR در اکثر موارد با بیان αβ و در موارد اندکی γδ قابل شناسایی است. جهشهای فعالکننده Notch1 اغلب در T-ALL یافت میشود و ژنهای SCL(TAL-1)، HOX11، LMO2 و LMO1 بهصورت شایع در T-ALL ناهنجار میگردند.
گرچه با مارکرهای سلولی میتوان انواع زیرگروههای T را تشخیص داد ولی ارتباط آنها با پیشآگهی دارای تناقض است. در بازنگری جدید به لوسمی حاد لنفوبلاستیک تحت عنوان Early T precursor ALL (ETP-ALL) به ایمونوفنوتایپ و ژنتیک پیشساز اولیه سلول T اشاره شده است که برخی ویژگیهای مایلوئیدی و سلول مادری را هنوز دارا است. سلولهای بلاست در ETP-ALL دارای بیان CD7 و مثبت بودن برای یک یا بیشتر از مارکرهای CD34، CD117، HLA-Dr، CD13، CD133، CD11b و CD65 بوده که مربوط به رده مایلوئیدی/ مادری میباشند. سلولهای سرطانی برای مارکر CD2 و CD3 سیتوپلاسمی و گاهی CD4 مثبت هستند. جهشهای مایلوئیدی مانند Flt3، NRAS/KRAS و IDH1,2 با شیوع زیاد در ETP-ALL مشاهده میشود و پیشآگهی نامطلوب است.
لوسمیهای حاد لنفوبلاستیک رده B از نظر فنوتایپ به pro B، Early pre B، pre B و B-ALL طبقهبندی میگردند. لوسمیهای ALL با ادغام ژنتیکی t(12;21) (ETV6-Runx1) حدود 25% لوسمی لنفوبلاستیک در کودکان با پیشآگهی مطلوب و پاسخ بسیار مطلوب به درمان آسپارژیناز همراه است. لوسمی حاد لنفوبلاستیک با ادغام ژنهای E2A-PBX1 در جابهجایی t(1;19) مرفولوژی لنفوسیتهای pre B دارد و نتیجهی درمان رضایتبخش است. جابهجایی انکوژن E2A در جابهجایی t(17;19) با هایپرکلسمی و اختلال انعقادی همراه است. لوسمی حاد لنفوبلاستیک با جابهجایی ژن MLL (11q23) مانند t(4;11) در کودکان زیر یک سال با پیشآگهی نامطلوب، سلولهای نارس pro B، بیان مارکرهای مایلوئیدی و افزایش جرم تومور (tumor load) همراهی دارد.
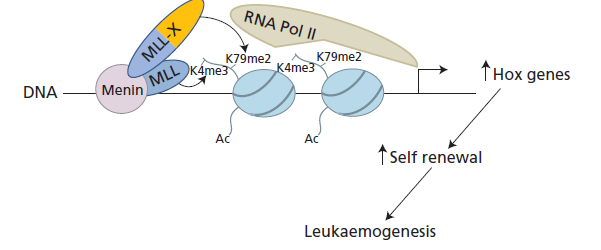
جابهجایی ژن MLL با افزایش بیان ژنهای خانواده HOX موجب سرطانهای لنفوبلاستی و مایلوبلاستی میشود
توضیحات تکمیلی
- ژن ASXL1 (additional sex combs like 1) روی کروموزوم 20 است که فرآورده پروتئینی آن نقش فاکتور نسخهبرداری برای بیان و سرکوب برخی ژنها را دارد. جهش در این ژن با اختلالات خون مانند سندرمهای مایلو دیسپلاستیک، لوسمی مایلومونوسیتیک مزمن و… همراهی دارد.
- ژن SF3B1 (splicing factor 3b subunit1) زیرواحد یک از فاکتور splicing کمپلکس پروتئینی 3b را شکل میدهد. جهش این ژن در سندرمهای مایلودیسپلاستیک بهویژه با سیدروبلاستهای حلقوی مشاهده شده است.
- ژن TET2 (Tet methylcytosine dioxygenase) که فرآورده آن کاتالیزکننده تبدیل متیل سیتوزین به 5 هیدروکسی متیل سیتوزین است، نقش کلیدی در دمتیلاسیون DNA دارد و روی کروموزوم 4q است. جهش این ژن با اختلالات مایلوپرولیفراتیو و مایلودیسپلاستیک همراهی دارد.
- ژن SRSF2 (serin arginin rich splicing factor 2) که فرآورده آن بخشی از spliceosome است، نقش مهمی در ارسال mRNA به سیتوپلاسم دارد. ژن بر روی کروموزوم 17 قرار دارد.
- ژن DNMT3A (DNA methyl transferase 3 alpha) نقش مهمی در متیلاسیون DNA دارد. جهش تکرارشونده این ژن در لوسمیهای AML و سندرمهای MDS مشاهده شده است. ژن روی کروموزوم 2p قرار دارد. انتقال گروه متیل به آرژینین روی هیستون از وظایف مهم فرآورده پروتئینی این ژن است.
- ژن U2AF1 (U2 small nuclear RNA auxillary factor 1) به گروه splicing factor خانواده SR تعلق دارد و یکی از چند اسپلایسئوزوم (spliceosome) است که اغلب در اختلالات خون جهش مییابد و جهش آن در سندرمهای مایلودیسپلاستیک با افزایش خطر تبدیل به لوسمی حاد همراه است. ژن روی کروموزوم 21 قرار دارد.
- ژن EZH2 (enhancer of zeste homolog 2) نقش لیزین متیل ترانسفراز بهویژه روی هیستونهای H3 و H4 دارد و ازاینرو در شیوه بیان ژنها نقش دارد. ژن روی کروموزوم شماره 7 قرار دارد. این ژن رمزدهنده، عضوی از خانواده polycomb است.
- ژن IDH1 (isocitrate dehydrogenase) که فرآورده آن کاتالیزکننده ایزوسیترات به 2-oxoglutarate است، جهش آن در برخی از بدخیمیهای خون مشاهده شده است. ژن روی کروموزوم شماره 2 قرار دارد.
- ژن Runx1 (Runt-related transcription factor 1) که فرآورده آن بهعنوان فاکتور نسخهبرداری اتصال مرکزی هستهای عمل میکند، برای خونسازی بسیار مهم است. فاکتور Runx1 با ایجاد کمپلکس با CBFβ، شکل فعال فاکتور نسخهبرداری را فراهم میکند. جهش Runx1 در لوسمیهای AML و اختلال پلاکتی مستعد به AML مشاهده شده است. جایگاه کروموزومی آن 21 است.
قدردانی: از همکاری خانم نیلوفر امیریان و خانم عالیه فاضلی، کارشناسان ارشد هماتولوژی علوم پزشکی تشکر میشود.
بازنگری لوسمیهای خانواده مایلوئیدی (WHO 2016) (1)
بازنگری لوسمیهای خانواده مایلوئیدی (2)
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام