بازنگری لوسمیهای خانواده مایلوئیدی (WHO 2016)
قسمت اول
دكتر حبیبالله گلافشان عضو هيئت علمي دانشگاه علوم پزشكي شيراز
نگين شكرگزار كارشناس ارشد هماتولوژی و بانک خون دانشگاه علوم پزشكي شيراز
تشخیص دقیقتر نئوپلاسمهای مایلوئیدی با بهکارگیری مرفولوژی سلول، سیتوژنتیک و بهویژه ژنتیک مولکولی که در سالهای اخیر دریچههای نوینی برای تشخیص گشوده است، منجر به بازنگری در طبقهبندی WHO این دسته از نئوپلاسمها در سال 2016 گردید.
لوسمی مزمن میلوسیتیک (CML)
لوسمی مزمن میلوسیتیک با مرفولوژی و آزمایش کروموزوم فیلادلفیا (جابهجایی کروموزوم 9 و 22 با ادغام BCR/ABL) بهراحتی قابل تشخیص بوده و در برهه زمانی دسترسی به بازدارندههای تیروزین کیناز (TKI) حتی طول عمر طبیعی را با پیگیری وسعت سلولهای +ABL/BCR، بررسی جهشهای جدید و مقاومت دارویی میتوان انتظار داشت. گفتنی است که درمان با بازدارندههای تیروزین کیناز، شیوع فاز شتابان (accelerated phase) این لوسمی را کاهش داده است. فاز شتابان لوسمی با تغییرات هماتولوژیکی، مرفولوژی، رخ دادن جهشهای جدید و نیز مقاومت به بازدارندههای تیروزین کیناز بهعنوان ایدهای در دست بررسی میباشد. فاز بلاستیک CML هنوز به حضور حداقل 20% از سلولهای بلاست در مغز استخوان یا خون محیطی و یا تجمع غده مانند سلولهای بلاست در خارج از فضای مغز استخوان (سارکوم اکسترامدولاری) نیاز دارد. بههرحال با توجه به اینکه فاز بحران بلاستیک از نوع لنفوبلاستیک در لوسمی مزمن میلوسیتیک ممکن است کاملاً ناگهانی باشد، از این رو یافتن لنفوبلاست در خون محیطی یا مغز استخوان نیاز به اقدام سریع با آزمایشهای ژنتیکی یا مولکولی برای تبدیل لوسمی به فاز لنفوبلاستیک دارد.
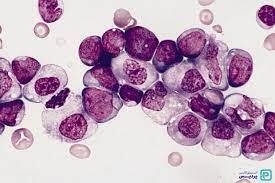
لوسمی مزمن مایلوسیتیک در فاز پایدار با سری کامل مایلوئیدی از مایلوبلاست تا نوتروفیل بالغ با بازوفیلی یا ائوزینوفیلی همراهی دارد. در مرحله پایدار، درصد سلولهای نوتروفیل و مایلوسیت بیشتر از سلولهای دیگر است. معمولاً مجموع بلاست و پرومایلوسیت کمتر از 5 درصد است
معیارهای فاز شتابان لوسمی مزمن میلوسیتیک (CML)
حضور هرکدام یا بیشتر از معیارهای هماتولوژی/ سیتوژنتیک زیر و یا عدم پاسخ به بازدارندههای تیروزین کیناز نشانه ورود مرحله پایدار بیماری به فاز شتابان است:
- تداوم لکوسیتوز (>100×109) رفراکتوری یا بدون پاسخ به درمان
- تداوم بزرگی طحال یا بزرگتر شدن آن و بدون پاسخ به درمان
- تداوم ترومبوسیتوز (>1000×109/L) بدون پاسخ به درمان
- تداوم کاهش پلاکتها (<100×109/L) بدون ارتباط به درمان
- حضور 20% یا بیشتر از سلولهای بازوفیل
- حضور 10 تا 19 درصد بلاست در خون محیطی یا مغز استخوان
- پیدایش اختلالات کروموزومی دیگر در سلولهایی که در بدو تشخیص از نظر کروموزوم فیلادلفیا مثبت بودهاند از قبیل دوبل شدن کروموزوم فیلادلفیا، تریزومی 8، ایزوکروموزوم 17q، تریزومی 19، کاریوتایپ پیچیده و غیرطبیعی شدن کروموزوم 3q
معیارهای پاسخ به بازدارندههای تیروزین کیناز (provisional response to TKI)
- مقاومت به اولین داروی بازدارنده تیروزین کیناز یا نبود پاسخ هماتولوژی کامل
- هرگونه نشانه هماتولوژی، سیتوژنتیک یا مولکولی که بیانگر مقاومت به دو سری پشت سرهم از داروهای بازدارنده باشد.
- رخداد دو یا بیشتر از جهشهای BCR/ABL در طی درمان با بازدارنده
گفتنی است که لوسمی مزمن میلوسیتیک در فاز پایدار با لکوسیتوز، افزایش مایلوسیت و نوتروفیل (دو موج افزایش)، مجموع بلاست و پرومایلوسیت کمتر از 5 تا 10 درصد و بازوفیلی یا ائوزینوفیلی نمایان میشود و مرحله شتابان، اخطاری جهت ورود بیماری به مرحله بلاستیک است.
با توجه به اینکه سرطانی شدن در سطح سلول مادر رخ داده است، از این رو فاز بلاستیک بهصورت لوسمیهای گوناگون میلوبلاستیک، مونوبلاستیک، مگاکاریوسیتیک و لنفوبلاستیک ظاهر میشود.
پاسخ هماتولوژیکی کامل در CML به حالتی گفته میشود که WBC<10000 و Plt<450000 در میلیمتر مکعب بوده و در شمارش افتراقی، گرانولوسیتهای نارس مشاهده نشود و یا طحال قابل لمس نباشد. با یافتن هر تعداد لنفوبلاست حتی کمتر از 10% بایستی تبدیل به فاز بلاستیک لنفوبلاستیک را با روشهای مولکولی تأئید کرد. حضور بیش از 20 درصد یا بیشتر از بلاستهای نوع دیگر یا تجمع بلاست بهصورت سارکوم یا کلروما در فضای خارج از مغز استخوان نشانه تبدیل به مرحله بلاستیک است. خوشههای بزرگ از مگاکاریوسیتهای کوچک و غیرطبیعی با افزایش فیبروز رتیکولین یا کلاژن در بیوپسی مغز استخوان نیز شاهدی بر فاز شتابان CML است که اغلب با یک یا چند مورد از معیارهای فاز شتابان همراه است.
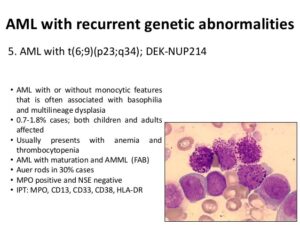
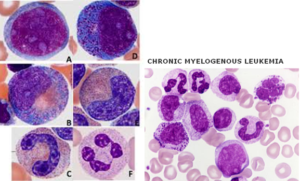
در تصویر فوق لوسمی مزمن مایلوسیتیک در فاز شتابان و یا لوسمی حاد مایلوبلاستیک با بازوفیلی در همراهی با جابجایی t (6;9) مشاهده میگردد
یافتههای جدید مولکولی و ژنتیکی، نیاز به بازنگری نئوپلاسمهایی که از نظر کروموزوم فیلادلفیا منفی هستند (BCR/ABL– MPNs) را یادآوری میکند؛ برای مثال:
1. جهش در پروتئین کالرتیکولین (CARL) که نقش چاپرون را ایفا میکند، علاوه بر جهشهای Jak2 و MPL یادآور تکثیر تک دودمانهای یا کلونالیتی (clonality) بوده و در پیشآگهی نقش دارد.
2. جهش در گیرنده G-CSF (CSF3R) همراهی چشمگیری با لوسمی مزمن نوتروفیلیک دارد.
لوسمی مزمن نوتروفیلیک (CNL)
از معیارهای پیشین WHO برای تشخیص لوسمی مزمن نوتروفیلیک، لکوسیتوز ≥25×109/L ، نوتروفیل/ باند ≥80%، کمتر از 10 درصد سری نابالغ مایلوئیدی و کمتر از یک درصد بلاست در خون محیطی بدون مرفولوژی دیسگرانولوپویز از قبیل اشکال پلگر و نوتروفیلهای هایپوگرانولار و بدون افزایش مونوسیت <1×109/L است. آزمایش مغز استخوان در این لوسمی شامل افزایش رده نوتروفیلی و کمتر از 5 درصد بلاست در میان جمعیت سلولهای هستهدار مغز استخوان است، ولی جهش در ژن CSF3R بهویژه جهش T618I که یک جهش فعالکننده است برای لوسمی CNL اختصاصی است؛ حتی در مواردی که درجه لکوسیتوز کمتر از حد معیار باشد. در غیاب جهش CSF3R، نوتروفیلی مداوم (حداقل سه ماه)، بزرگ بودن طحال و نبود علتی برای نوتروفیلی واکنشی از قبیل تومورهای پلاسماسل و برخی تومورهای دیگر، احتمال لوسمی داده میشود.
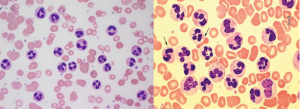
در لوسمی مزمن نوتروفیلیک، لکوسیتوز با غالب بودن سلولهای نوتروفیل بالغ و کمتر از 10 درصد سری نارس مایلوئیدی مشاهده میشود. جهش در گیرنده G-CSF در اکثر موارد این لوسمی را تشخیص میدهد
- با توجه به عدم موفقیت در تشخیص مواردی از پرخونی ورا در گذشته، با کم کردن میزان آستانه هموگلوبین و تقلیل آن از >18.5 به >16.5 برای آقایان و بیشتر از 16 برای خانمها ممکن است بتوان پلیسیتمی نهفته ورا (masked P.V) را زودتر تشخیص داد.
در این بازبینی به هیستولوژی مغز استخوان به علت تکرارپذیر بودن یافتههای تشخیصی اهمیت بیشتری داده شده است و در گروه معیارهای اصلی به شرح زیر قرار گرفته است:
معیارهای اصلی پرخونی ورا
- افزایش هموگلوبین >16/5 برای آقایان و >16 برای خانمها یا افزایش جرم گلبولهای قرمز (RCM)
- بیوپسی مغز استخوان که افزایش سلولاریته را با توجه به سن نشان میدهد، مرفولوژی پانمایلوز (panmyelosis) در پرخونی ورا است؛ به این مفهوم که هایپرپلازی سه ردهای (Trilineage) در ردههای اریتروئیدی، گرانولوسیتی و مگاکاریوسیتی مشاهده میشود. افزایش مگاکاریوسیتهای بالغ با اشکال گوناگون (pleomorphic) در بیوپسی مغز استخوان قابل توجه است. نمای پانمایلوز بهصورت افزایش همزمان هموگلوبین و گلبولهای سفید و پلاکت در خون محیطی درمیآید.
- حضور جهش Jak2V617F یا جهشهای دیگر ژن Jak2 در اگزون 12
معیار فرعی: سطح اریتروپویتین کمتر از نرمال
گرچه برای بیمار پرخون با هموگلوبین بیشتر از 18/5 (HCT 55.5%) برای آقایــــــــــان یا 16.5 (HCT 49.5%) برای خانمها و با حضور جهش Jak2 ممکن است ارزیابی مغز استخوان نیاز نباشد، ولی از آنجا که مایلوفیبروز اولیه در مراحل ابتدایی حتی در 20 درصد موارد پرخونی ورا مشاهده شده است، از این رو بیوپسی مغز استخوان در پیشبینی تبدیل پرخونی به مایلوفیبروز دارای ارزش است. تشخیص افتراقی ET (ترومبوسیتمی اولیه) از مایلوفیبروز اولیه در مراحل اولیه (Pre PMF) بسیار مهم است.
طبقهبندی نئوپلاسمهای مایلوئیدی در سال 2008 توسط WHO از کشف جهشهای Jak2 (Chr 9p24) یا V617F در اگزون شماره 14 ژن ژانوس کیناز (Jak) و جهشهای حذف یا اضافه شدن نوکلئوتید (indels) در اگزون 12 ژن Jak2 و نیز جهش در ژن MPL (گیرنده ترومبوپویتین) روی کروموزوم 1p34 بهویژه در رمز W515 بهعنوان معیارهای اصلی تشخیص بهره میبرد. جهش Jak2V617F در حدود 95% موارد پرخونی ورا و حدود 60 درصد موارد ET و PMF (مایلوفیبروز اولیه) مشاهده میگردد.
حذفها (deletion) و اضافه شدن (insertion) مختلف بهویژه در کدونهای 543-537 اگزون 12 ژن Jak2 در حدود 3 تا 5 درصد موارد پرخونی ورا که در آنها جهش Jak2V617F منفی است با روشهای حساس قابل شناسایی است. در حدود 4 تا 8 درصد موارد ترومبوسیتمی اولیه و مایلوفیبروز، جهش در اگزون 10 ژن MPL بهویژه در رمز 515 با شیوع جایگزینی R، A، L، K W و بهندرت با جایگزینی در رمز 505 (S>N) مشاهده میشود.
در سال 2013 جهش در ژن کالرتیکولین (CARL) روی کروموزوم 19p13.2 که پروتئین آن نقش چاپرون در رابطه با شبکه اندوپلاسمیک دارد، در بیماران مبتلا به ترومبوسیتمی اولیه و مایلوفیبروز که از نظر جهشهای Jak2/MPL منفی بودند گزارش گردید و تا حد زیادی شکاف تشخیص مولکولی ET و PMF را پر کرد.
جهشهای CARL، حذفها و اضافه شدنهای (indels) هتروژن است که تمامی آنها در اگزون شماره 9 که قسمت انتهایی (C-terminal) پروتئین را رمزدهی میکند، رخ داده است. دو جهش شایع آن که شیوعی بیشتر از 85 درصد دارد، تایپ یک (حذف 52 جفت باز a52bp deletion P.L367F*s46) و تایپ 2 (اضافه شدن 5 جفت باز a 5 bp insertion P.K385fs*47) میباشد، در حالی که بقیه جهشها بهعنوان شبهتایپ یک و شبهتایپ دو طبقهبندی شدهاند.
جهشهای تایپ 2 کالرتیکولین (CARL) ترجیحاً با ترومبوسیتمی اولیه (ET) و جهشهای تایپ یک بهطور غالب مایلوفیبروز (PMF) را همراهی میکند. جهشهای یادشده Jak2V617F و اگزون 12 بهعنوان معیارهای اصلی در تشخیص پلیسیتمی ورا و جهشهای Jak2V617F و CARL و MPL برای تشخیص ET و PMF با توجه به بازنگری طبقهبندی نئوپلاسمهای مایلوئیدی در سال 2016 میباشند، از این رو تشخیص به روز نئوپلاسمهای مایلوئیدی نیاز به دانستن جهشها دارد. بههرحال وقتی که جهشهای فوق در نمونههای تشخیصی منفی باشد، میتوان از معیارهای فرعی WHO برای حمایت تشخیصی استفاده کرد.
تعدادی از بیماران مبتلا به پرخونی ورا که از نظر جهشهای Jak2 منفی هستند ممکن است دارای جهشهای دیگر در ژنهایی مانند SH2B3/LNK باشند. از طرف دیگر حدود 20% از موارد ET و 10 تا 15% از موارد PMF که فاقد جهشهای اصلی (Jak2, CARL, MPL) هستند، تحت عنوان PMF و ET با سهتایی منفی (Triple negative) از آنها یاد میشود. بازنگری 2016 برای بیماران مبتلا به مایلوفیبروز که برای هر سه جهش اصلی منفی هستند، از جستوجو برای جهشهای دیگر (Nondriver mutations) از قبیل جهش در ژنهای ASXL1، EZH2، TET2، IDH1/IDH2، SRSF2 و SF3B1 که میتوانند مارکر کلونالیتی باشند، حمایت میکند. گرچه این جهشها اختصاصی و انحصاری برای یک لوسمی نیستند ولی در حدود 50 درصد از موارد مایلوفیبروز مشاهده شدهاند و حتی با وسعت دادن پانل تشخیصی تا 81 درصد موارد دارای حداقل یک مارکر کلونال بودهاند.
خطر ترومبوز در ET ارتباط چشمگیری با جهشهای Jak2 دارد، درحالی که حضور جهش تایپ یک CARL پیشآگهی بهتر مایلوفیبروز را دربر دارد.
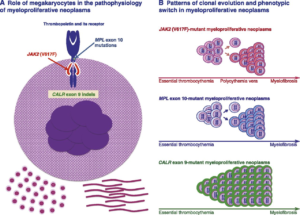
جهشهای کالرتیکولین و Jak2 و MPL از مهمترین معیارهای تشخیصی احتمالات مایلوپرولیفراتیو هستند که از نظر کروموزوم فیلادلفیا منفی هستند
معیارهای تشخیصی مایلوفیبروز در فاز pre PMF و مایلوفیبروز واضح (overt)
WHO Criteria
| Pre – PMF | overt PMF |
| معیار اصلی | معیار اصلی |
| 1) تکثیر مگاکاریوسیتها در مغزاستخوان همراه با مورفولوژی آتیپیک و بدون فیبروز بیشتر از درجه یک
2) افزایش سلولاریته با هایپرپلازی گرانولوسیتها 3) حضور یکی از جهشهای Jak2، CARL، MPL و کنار گذاشتن نئوپلاسمهای دیگر مایلوئیدی. در غیاب جهشهای فوق حضور یکی از جهشهای کلونال مانند ASXL1، EZH2، IDH1/IDH2 و کنار گذاشتن فیبروز به علت واکنشی |
1) تکثیر مگاکاریوسیتها با مورفولوژی آتیپیک و فیبروز رتیکولین/کلاژن درجه 2 و 3
2) کنار گذاشتن اختلالات دیگر مایلوپرولیفراتیو با معیارهای WHO 3) حضور یکی از جهشهای Jak2، CARL و MPL و در صورت منفی بودن حضور یک یا چند از جهشهای کلونال |
| معیارهای فرعی | معیارهای فرعی |
| 1) کمخونی
2) لکوسیتوز ≥11000 در میلیمتر مکعب 3) طحال قابل لمس 4) افزایش سطح LDH بالاتر از نرمال |
1) کمخونی
2) لکوسیتوز >11000 3) طحال بزرگ 4) افزایش LDH 5) واکنش لکواریتروبلاستیک خون محیطی |
سه معیار اصلی همراه با حداقل یک معیار فرعی، تشخیص را قطعی میکند. درجه فیبروز مغز استخوان از MF0 تا MF3 درجهبندی میشود.
توضیح: فیبروز مغز استخوان ثانویه به عفونت، اختلالات اتوایمیون، بیماریهای التهابی، لوسمی سلولهای مویی و تومورهای متاستاتیک بهصورت واکنشی ممکن است مشاهده شود.
واکنش لکواریتروبلاستیک به مفهوم حضور همزمان سری نارس مایلوئیدی و گلبولهای قرمز هستهدار همراه یا بدون گلبولهای قطره اشکی است.
معیارهای تشخیصی ترومبوسیتمی اولیه (ET)
معیارهای اصلی:
- شمارش پلاکت ≥450000 در میلیمتر مکعب
- حضور یکی از جهشهای Jak2، CARL یا MPL
- تکثیر چشمگیر رده مگاکاریوسیتها در مغز استخوان با افزایش تعداد مگاکاریوسیتهای بالغ هایپرلوبوله، افزایش غیرقابل توجه یا میل به چپ ردههای نوتروفیلی و اریتروئیدی و بهندرت فیبروز رتیکولین درجه یک
- کنار گذاشتن اختلالات دیگر مایلوپرولیفراتیو و سندرمهای مایلودیسپلاستیک با معیارهای WHO
معیارهای فرعی:
حضور یک نشانه تکثیر کلونال و کنار گذاشتن ترومبوسیتوز واکنشی
تشخیص به هر 4 معیار اصلی یا سه معیار اصلی با یک معیار فرعی نیاز دارد.
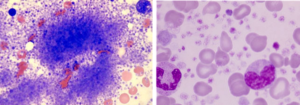
در ترومبوسیتمی اولیه شمارش پلاکت از بیشتر از 500000 تا میلیونها در میلیمتر مکعب متغیر است
اختلالات ژنتیکی مولکولی در نئوپلاسمهای مایلوئیدی/ لنفوئیدی همراه با افزایش ائوزینوفیل
تکثیر کلونال ائوزینوفیل در بازآرایی ژنهای گیرنده رشد پلاکتی PDGFRA/B بر روی کروموزومهای 4 و 5 و بازآرایی در گیرنده فاکتور رشد فیبروبلاست روی کروموزوم 8 (FGFR1) و فیوژن ژنهای PCM1-Jak2 رخ میدهد که با نئوپلاسمهای سلولهای B یا T و یا AML همراهی دارند.
نئوپلاسمهای مایلوئیدی/ لنفوئیدی با ائوزینوفیلی
| ژن | تظاهر | ژنتیک | پاسخ به بازدارنده تیروزین کیناز |
| PDGFRA | ائوزینوفیلی، افزایش تریپتاز سرمی و افزایش ماست سل در مغز استخوان | حذف بینابینی 4q با ادغام Fip1L1-PDGFRA و حداقل 60 انتخاب دیگر برای ادغام | مثبت |
| PDGFRB | ائوزینوفیلی و افزایش مونوسیت شبیه به لوسمی مزمن میلومونوسیتیک (CMML) | t(5;12) ETV6/PDGFRB با حداقل 25 انتخاب دیگر برای ادغام | مثبت |
| FGFR1 | ائوزینوفیلی اغلب همراه با T-ALL یا AML | جابهجایی 8p با انتخابهای متعدد ژنی | بدون پاسخ |
| PCM1-Jak2 | ائوزینوفیلی، بهندرت با نـــمای T-LBL یــا B-ALL
مغز استخوان میل به چپ در سری اریتروئیدی و تجمع لنفوسیتی را نشان میدهد |
t(8;9) PCM1/Jak2 | امکان پاسخ |
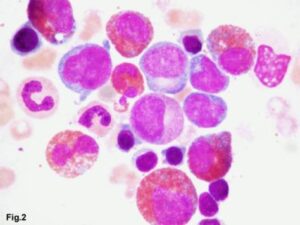
در علت شناسایی ائوزینوفیلی بایستی همراهی آن با لوسمیهای مایلوئیدی یا لنفوئیدی را مدنظر قرار داد. در تصویر فوق لوسمی مزمن ائوزینوفیلیک همراه با بلاست مشاهده میگردد
بازآرایی و ادغام ETV6/Jak2 و BCR/Jak2 در اکثر موارد با نمای B-ALL بروز میکنند. افزایش تک دودمانه مونوکلونال ائوزینوفیل که در قالب ژنتیکی فوق قرار نگیرد، تحت عنوان CEL-NOS (لوسمی ائوزینوفیلی مزمن) نام میگیرد که برای تشخیص نیاز به شمارش مطلق ≥1.5×109/L ائوزینوفیل در خون محیطی و بیشتر از 2 درصد میلوبلاست در خون محیطی یا 5 تا 19 درصد از سلولهای بلاست در مغز استخوان دارد. سیتوژنتیک غیرطبیعی در اکثر موارد تریزومی 8، t(10;11) و t(7;12) بوده و برخلاف نئوپلاسمهای همراه با ائوزینوفیلی از قبیل PDGFRA/B به بازدارندههای تیروزین کیناز پاسخ نمیدهند.
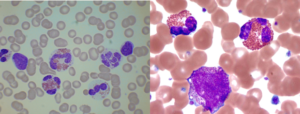
در تصویر فوق ائوزینوفیل همراه با سلولهای نارس مایلوئیدی مشاهده میشود. اختلالات کروموزومی 4 و 5 و 8 شایعترین علت ائوزینوفیلی کلونال هستند
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام