بازنگری لوسمیهای خانواده مایلوئیدی (WHO 2016)
قسمت دوم
دكتر حبیبالله گلافشان عضو هيئت علمي دانشگاه علوم پزشكي شيراز
نگين شكرگزار كارشناس ارشد هماتولوژی و بانک خون دانشگاه علوم پزشكي شيراز
سندرمهای مایلودیسپلاستیک (Myelodysplastic syndromes)
سندرمهای مایلودیسپلاستیک گروهی از نئوپلاسمهای تک دودمانه (کلونال) بوده که با خونسازی غیرمؤثر (ineffective) همراه هستند و استعداد تبدیل به لوسمی حاد مایلوبلاستیک را دارند. نمای خونسازی غیرمؤثر بهصورت تغییرات دیسپلاستیک (morphologic dysplasia) در یک یا تمام ردههای سلولی ظاهر شده و افزایش آپوپتوز مورفولوژی ناهنجار سلولهای خون منجر به سایتوپنی تک ردهای (single lineage) یا تمام ردهای (multilineage) میشود.
طبقهبندی WHO بهطور ویژه بر روی تغییرات دیسپلاستیک مرفولوژی و درصد سلولهای بلاست تکیه دارد.
از مهمترین مرفولوژیهای دیسپلاستیک رده گرانولوسیتی میتوان به مرفولوژی پلگر کاذب (psudopelger)، نوتروفیـــلها با کاهش گرانــــــول، نوتروفیـــــــل با هسته حلقوی و هایپرسگمانته شدن بیقواره (Bizarre hypersegmentation) اشاره کرد.
سلولهای مونوسیت که افتراق آنها از مایلوسیت مشکل است (paramyeloid) از دیگر مرفولوژیهای دیسپلاستیک میباشند. حضور میکرومگاکاریوسیت از مرفولوژیهای نسبتاً اختصاصی دیسپلاستیک است. اشکال مختلف گلبولهای قرمز بهویژه ماکروسیت و دایمورف ممکن است در گستره محیطی مشاهده شود. مغز استخوان ردههای اریتروئیدی را با تغییرات مگالوبلاستوئید، هستههای شکسته، لوبوله و چندهستهای نشان میدهد.
برای گزارش تغییرات دیسپلاستیک در یک رده بایستی حداقل 10% از سلولهای آن رده دارای تغییرات دیسپلاستیک باشند. سایتوپنی شامل Hb<10، شمارش مطلق نوتروفیل کمتر از 1800(با توجه به نژاد) و شمارش پلاکت کمتر از 100000 در میلیمتر مکعب است.
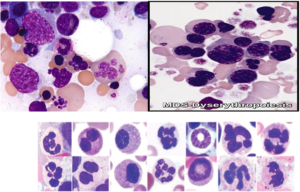
تصاویر ردیف بالا تغییرات دیسپلاستیک گلبولهای قرمز هستهدار از قبیل چندهستهای، هستهی شکسته و ناهمگنی رشد هسته و سیتوپلاسم را نشان میدهد. تصاویر ردیف پایین نشاندهندهی تغییرات دیسپلاستیک در نوتروفیلها با هسته حلقوی و پلگر کاذب میباشند
سیدروبلاستهای حلقوی (MDS-RS)
مورفولوژی سیدروبلاست حلقوی برای تشخیص زیرگروههایی از سندرمهای مایلودیسپلاستیک الزامی است. در سیدروبلاست حلقوی، گرانولهای آبی آهن، مانند یقه پیرامون هسته را فرا میگیرد که با رنگآمیزی پرل قابل مشاهده است. جهش در ژن SF3B1 (spliceosome) عامل تولید سیدروبلاستهای حلقوی است؛ برای مثال سندرم مایلودیسپلاستیک با سیدروبلاستهای حلقوی MDS-RS دارای جهش در ژن اسپلایسیزوم (spliceosome) SF3B1 میباشند و از این رو با تأیید این جهش دیگر نیازی به مشاهده حداقل 15% سیدروبلاست حلقوی در میان جمعیت گلبولهای قرمز هستهدار برای تشخیص نیست و حتی کمتر از 5% سیدروبلاست کفایت میکند.
با وجود اینکه سیتوژنتیک غیرطبیعی بهجز حذف –5q برای تشخیص گروهی ویژه از MDS اختصاصی نیست، اما همراهی چشمگیری با پیشآگهی دارد.
جهشهای تکرارپذیر در 80 تا 90% از بیماران مبتلا به MDS شبیه به نئوپلاسمهای مایلوئیدی، گزارش گردیده است که برای مثال میتوان به جهش در ژنهای SF3B1، TET2، SRSF2، ASXL1، DNM3A، Runx1، U2AF1، T53 و EZH2 اشاره کرد. گاهی جهــشهای کلونال شبیه به MDS در افراد سالخورده بدون ابتــــــــــــــــــــلا به MDS رخ میدهــــــد که تحت عنــــوان CHIP (clonal hematopoiesis of indetermined potential) یا هماتوپویز کلونال با پتانسیل نامشخص از آن یاد میشود.
اختلالات کروموزومی–y و del 20q در غیاب مرفولوژیهای خاص MDS بیانگر ابتلای قطعی به سندرمهای مایلودیسپلاستیک نیستند. شناسایی جهش P53 در بیماران MDS سفارش میشود زیرا بیانگر گروهی با پیشآگهی بسیار نامطلوب است.
کلیدواژهها در طبقهبندی سندرمهای مایلودیسپلاستیک
- MDS (Myelodysplastic Syndrome): سندرم مایلودیسپلاستیک
- SLD (Single Lineage Dysplasia): سندرم مایلودیسپلاستیک با دیسپلازی تک ردهای یا ناهنجاری تک ردهای. ناهنجاری مرفولوژی ممکن است شامل رده اریتروئیدی، مایلوئیدی و یا مگاکاریوسیتی باشد.
- MLD (Multilineage Dysplasia): سندرم مایلودیسپلاستیک با دیسپلازی چند ردهای که در این حالت یک تا دو تا سه رده سلولی دارای ناهنجاری مرفولوژی هستند.
- MDS-RS: سندرم مایلودیسپلاستیک با سیدروبلاستهای حلقوی با دیسپلازی تک ردهای (MDS-RS-SLD) یا چند ردهای (MDS-RS-MLD)
- MDS del (5q): سندرم مایلودیسپلاستیک با تک اختلال حذف 5q
- MDS EB: سندرم مایلودیسپلاستیک با افزایش شمارش بلاست که دو گروه EB1 با 2 تا 4% بلاست در خون محیطی و 5 تا 9% بلاست در مغز استخوان و EB2 با 10 تا 19% بلاست در مغز استخوان و 5 تا 19% بلاست در خون محیطی همراه بوده، آور راد مشاهده نگردیده و دیسپلازی ممکن است از صفر تا سه ردهای و سایتوپنی یک تا سه ردهای مشاهده گردد.
- MDS:U: سندرم مایلودیسپلاستیک که در طبقهبندی جای نمیگیرد و در این حالت خون محیطی تا یک درصد بلاست و مغز استخوان کمتر از 5 درصد بلاست بدون آور راد دارد. دیسپلازی ممکن است مشاهده شود یا تنها بر مبنای سیتوژنتیک تشخیص داده شود.
- Refractory cytopenia of children: سایتوپنی رفراکتوری کودکی با دیسپلازی و سایتوپنی و بدون سیدروبلاست حلقوی با کمتر از 2 درصد بلاست در خون محیطی و کمتر از 5 درصد در مغز استخوان
توجه داشته باشید که برای تشخیص MDS با سیدروبلاست حلقوی، حداقل نیاز به 15 درصد سیدروبلاست در گلبولهای قرمز هستهدار هست، اما چنانچه جهش SF3B1 مثبت باشد، میتوان به کمتر از 5% تقلیل داد. در بقیه موارد MDS، سیدروبلاست حلقوی ممکن است مشاهده شود یا نشود و معمولاً کمتر از 15% بوده و در این موارد جهش SF3B1 مشاهده نمیگردد.
هرگونه اختلال کروموزومی ممکن است در MDS مشاهده شود و تنها حالت حذف 5q است که در یک گروه اختصاصی جای میگیرد.
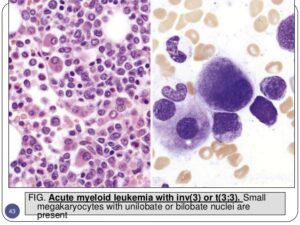
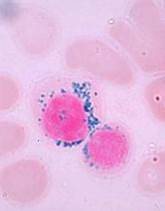
سندرم مایلودیسپلاستیک در زیرگروه 5q- با کمخونی، افزایش پلاکتها در خون محیطی و حضور فراوان میکرومگاکاریوسیتهای تک یا دو لوبه در مغز استخوان مشاهده میگردد
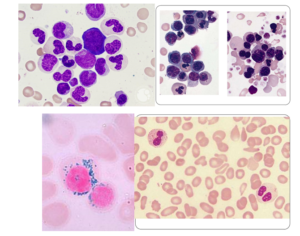
سندرمهای مایلودیسپلاستیک با دیسپلازی چند ردهای مشاهده میشود. سیدروبلاست حلقوی در بسیاری از سندرمهای مایلودیسپلاستیک مشاهده میشود. در این مرفولوژی گرانولهای آبی آهن، اطراف هسته نرموبلاست را احاطه میکند
نئوپلاسم های همپوش مایلوپرولیفراتیو/ مایلودیسپلاستیک (MDS/MPN)
این خانواده از بدخیمیها دارای بازنگری به شرح زیر است:
1- لوسمی مزمن مایلومونوسیتیک (CMML)
2- لوسمی مزمن میلوسیتیک اتیپیک (aCML)
3- MDS/MPN با سیدروبلاست حلقوی و ترومبوسیتوز (MDS/MPN RS-T)
4- لوسمی مایلومونوسیتیک جوانی (JMML)
کاریوتایپ غیرطبیعی شبیه به سندرمهای مایلودیسپلاستیک در بیماران مبتلا به سندرمهای همپوش ممکن است رخ دهد. در 80% موارد CMML، جهشهای SRSF2 و ASXL1/TET2 مشاهده میگردد. جهش ژنهای ASXL1 و NPM1 بیانگر تهاجمی بودن CMML است.
معیارهای تشخیصی لوسمی مزمن مایلومونوسیتیک (CMML)
1- تداوم مونوسیتوز ≥1×109/L با شمارش مونوسیت بیشتر از 10 درصد از گلبولهای سفید. مونوسیتوز برای حداقل سه ماه تداوم داشته و سایر علل مونوسیتوز کنار گذاشته شده باشد.
2- کمتر از 20% سلول بلاست در خون محیطی یا مغز استخوان
3- دیسپلازی یا مرفولوژی ناهنجار در یک یا چند رده سلولی از قبیل میکرومگاکاریوسیت در مغز استخوان یا مرفولوژی پلگر کاذب و هسته حلقوی و کاهش گرانولها در نوتروفیل و یا مشاهده گلبول هستهدار غیرطبیعی
4- حضور کمتر از 10% سلولهای نارس مایلوئیدی در خون محیطی
5- چنانچه CMML با افزایش ائوزینوفیل همراه شد، بایستی بازآرایی ژنهای PDGFRA، PDGFRB، FGFR1 و یا PCM/Jak2 را مدنظر داشت.
6- منفی بودن جهشهای BCR/ABL، CARL، Jak2 و MPL
7- شمارش بلاست شامل مایلوبلاست و یا مترادف با آن مونوبلاست و یا پرومونوسیت است. هستههای پیچخورده و تادار، سیتوپلاسم خاکستری، تعدادی گرانولهای پوستپیازی و کروماتین مخملی از ویژگیهای پرومونوسیت است. حضور جهش در ژنهای TET2، SRSF2، ASXL1 و SETBP1 ممکن است تشخیص CMML را قطعیتر کند.
جداسازی CMML به تایپ مایلوپرولیفراتیو با شمارش WBC≥13000 و به تایپ دیسپلاستیک با شمارش WBC<13000 در میلیمتر مکعب به نظر میرسد ناشی از انحراف علائمرسانی مسیر RAS/MAPKباشد.
از آن جا که تعداد بلاست بیانگر پیشآگهی بهتری در لوسمی است، از این رو لوسمی با کمتر از 2% بلاست در خون محیطی و کمتر از 5% در مغز استخوان بهصورت CMML-0 و با 2 تا 4% بلاست در خون محیطی و 5 تا 9% بلاست در مغز استخوان تحت عنوان CMML-1 و برای آن دسته از لوسمیها با 5 تا 19% بلاست در خون محیطی و 10 تا 19% بلاست در مغز استخوان یا تنها مشاهده آور راد (Auer Rod)، بهصورت CMML-2 از آن یاد میشود. برای تشخیص دقیقتر تعداد بلاستها، میلوبلاست، پرومونوسیت و مونوبلاست نیاز به فنوتایپ ایمونولوژیک یا فلوسیتومتری است.
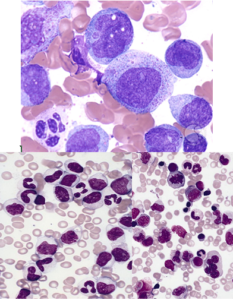
لوسمی مایلومنوسیتیک مزمن (CMML) با افزایش مونوسیتها، سلولهای بلاست و سلولهای نارس مایلوئیدی همراه با تغییرات دیسپلاستیک از قبیل پلگر کاذب و نوتروفیلهای کمگرانول تظاهر میکند
لوسمی مایلومونوسیتیک جوانی (JMML)
لوسمی مایلومونوسیتیک جوانی یک اختلال تهاجمی کلونال بافت خونساز با تکثیر سری گرانولوسیتی و مونوسیتی است که کودکان و بچهها را هدف قرار میدهد. حدود 90% بیماران حامل جهشهای سوماتیک یا جهش در سلولهای زایا (germ line) از قبیل PTPN11، KRAS، NRAS، CBL یا NF1 میباشند. این ناهنجاریهای ژنتیکی منجر به فعال کردن مسیر علائمرسانی RAS/MAPK میگردد.
معیارهای تشخیصی لوسمی مایلومونوسیتیک جوانی (JMML) از دیدگاه هماتولوژی و ژنتیک
1- شمارش مونوسیت خون محیطی ≥1×109/L
2- شمارش بلاست در خون محیطی یا مغز استخوان کمتر از 20%
3- بزرگی طحال
4- منفی بودن ادغام BCR/ABL
5- جهش سوماتیک در PTPN11 یا KRAS یا NRAS
6- تشخیص بالینی جهش NF1 یا جهش سلولهای زایا CBL

سندرم نونان با اختلالات اسکلتی و جهش در ژن PTPN11 شخص را مستعد ابتلا به لوسمی مایلومونوسیتیک جوانی میکند. مونوزومی 7 و نوروفیبروماتوز نیز از عوامل مستعدکننده به لوسمی جوانی مایلومونوسیتیک است
برای تشخیص ژنتیکی JMML، شناسایی یکی از جهشها کافی است و در صورت منفی بودن جهشها، موارد زیر بایستی صادق باشند:
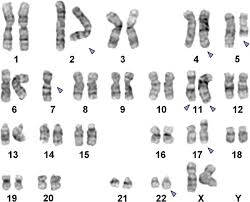
مونوزومی 7
- مونوزومی 7 یا اختلالات دیگر کروموزومی و یا حداقل دو مورد از موارد زیر:
- افزایش هموگلوبین F با توجه به سن
- حضور پیشسازهای اریتروئیدی یا مایلوئیدی در خون محیطی
- افزایش حساسیت کلونیهای خونساز به GM-CSF
- هایپرفسفوریلاسیون (hyperphosphorylation) STAT5
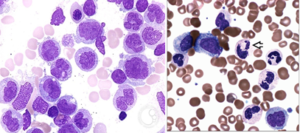
لوسمی مایلومونوسیتیک جوانی شبیه به نمای لوسمی مزمن مایلومونوسیتیک مزمن تظاهر کرده و با افزایش هموگلوبین F همراه میگردد
لوسمی مزمن مایلوسیتیک اتیپیک (aCML)
لوسمی مزمن مایلوسیتیک اتیپیک در زیرگروه MDS/MPN قرار دارد. امروزه افتراق این لوسمی از لوسمی مزمن نوتروفیلیک (CNL) که با نوتروفیلی تظاهر میکند، آسان شده است.
با وجود اینکه لوسمی مزمن نوتروفیلیک همراهی چشمگیری با جهش CSF3R دارد، این جهش تنها در کمتر از 10% از موارد aCML مثبت است و از طرف دیگر جهشهای SETBP1 و یا ETNK1 در حدود 30% موارد aCML مشاهده میشود. جهشهای Jak2، CARL و MPL (MPN driver mutation) همگی در aCML منفی هستند.
معیارهای تشخیصی لوسمی مزمن مایلوسیتیک اتیپیک (aCML)
1- لکوسیتوز خون محیطی همراه با رده نابالغ آن (سلولهای پرومایلوسیت، مایلوسیت و متامایلوسیت ≥10% از لکوسیتها)
2- دیسگرانولوپویز که علاوه بر اشکال پلگر ممکن است فشردگی غیرطبیعی کروماتین را نشان دهد.
3- افزایش حداقلی بازوفیل (کمتر از 2 درصد) یا نبود آن
4- افزایش حداقلی مونوسیت (کمتر از 10 درصد) یا نبود آن
5- مغز استخوان هایپرسلولار با دیسپلازی گرانولوسیتی همراه یا بدون همراهی با دیسپلازی اریتروئیدی و مگاکاریوسیتی
6- کمتر از 20% بلاست
7- منفی بودن جهشهای تشخیصی برای ET، PV، PMF و CML
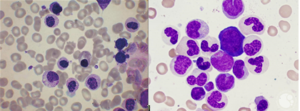
لوسمی مزمن مایلوسیتیک اتیپیک (aCML) شبیه به لوسمی مزمن مایلوسیتیک کلاسیک است با این تفاوت که تغییرات دیسپلاستیک مشاهده گردیده و بازوفیلی و افزایش پلاکت چشمگیر نیست
بازنگری لوسمیهای خانواده مایلوئیدی (WHO 2016) (1)
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام