سندرم اهلرز- دانلوس
Ehlers–Danlos syndrome
شاهین اسعدی*،الناز زرگری ( دانشجویان ژنتیک مولکولی)، دکتر سعید قربیان متخصص ژنتیک

سندرم اهلرز- دانلوس یک اختلال بافت همبند در انسان است که بصورت ژنتیکی به ارث می رسد. بقراط 400 سال قبل از میلاد به ویژگیهای سندرم EDS اشاره کرده بود اما اولین بار توسط دو پزشک بنام ادوارد اهلرز از دانمارک و هانری اکساندر دانلوس از فرانسه در قرن 20 گزارش شد. این اختلال توسط نقص در ساختار تولید و یا پردازش پروتئین کلاژن در اثر جهش در ژن COL5A یا COL3A ایجاد میشود. کلاژن یکی از پرکاربردترین پروتئینهای بدن میباشد که حدود 25 درصد از کل پروتئینهای انسانی را تشکیل میدهد و بعنوان بافت گرانوله فیبر مشبک ایفای نقش میکند که توسط فیبروبلست جوان تولید میشود.
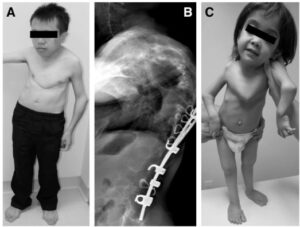
کلاژن همچنین در دیواره شریانها، پوست، روده و رحم یافت میشود و عامل مهم در قدرت فیزیکی پوست، مفاصل، عضلات، لیگامانها، رگهای خونی و ارگانهای احشایی میباشد. در سال 1997 ژنتیکدانان شش نوع اصلی از سندرم اهلرز- دانلوس را شناسایی کردند. در جدول زیر شش نوع اصلی سندرم اهلرز- دانلوس با ویژگیهای مربوطه مشخص شده است:
| ژن | ویژگی | شماره | نام |
| COL3A1،TNXB | شیوع 1 در 10000 تا 15000 تولد که توسط اتوزومال مغلوب یا غالب به ارث میرسد. بیماران با تجربه درد مزمن اسکلتی عضلانی و شل شدگی پوست و در رفتگی مفاصل مواجه هستند.
|
Type 3 | Hypermobility |
| COL5A1،COL5A2،COL1A1 | شیوع 1 در 20000 تا 50000 تولد که فقط توسط اتوزومال غالب به ارث میرسد. بیماران با اختلال متوسط تا شدید شلی پوستی و مفاصل موجه هستند.
|
Type 1،2 | Classical |
| COL3A1 | شیوع 1 در 100000 تا 250000 تولد که با الگوی اتوزومال غالب به ارث میرسد. نوع عروقی به عنوان شدیدترین مشخصه سندرم اهلرز-دانلوس محسوب میشود به دلیل پارگی رگهای خونی در ارگانها
|
Type 4 | Vascular |
| PLOD1 | شیوع تاکنون 60 تولد در جهان که توسط الگوی اتوزومال مغلوب به ارث میرسد. بیماران با ضعف شدید عضلانی و انحنای ستون فقرات مواجه هستند.
|
Type 6 | Kyphoscoliosis |
| COL1A1،COL1A2 | شیوع تاکنون 30 تولد در جهان که توسط الگوی اتوزومال غالب به ارث میرسد. بیماران با شلی شدید مفاصل و دررفتگی هر دو باسن مواجه هستند.
|
Type 7 A،B | Arthrochalasia |
| ADAMTS2 | شیوع تاکنون 10 تولد در جهان که توسط الگوی اتوزومال مغلوب به ارث میرسد. بیماران با افتادگی پوست شدید و شکنندگی استخوانها مواجه هستند.
|
Type 7 C | Dermatosparaxis |
بطور کلی آمار تحقیقاتی از سندرم اهلرز- دانلوس نشان می دهد که شیوع این بیماری 1 در 2500 تا 5000 تولد میباشد.
علائم و نشانه ها :
علائم به طور گسترده اما با کمی تفاوت در هر کدام از انواع سندرم EDS وجود دارد. در هر کدام از انواع سندرم EDS کاهش میزان پروتئین کلاژن و معیوب بودن آن از لحاظ نحوه ساختار دیده می شود.

اسکلتی عضلانی :
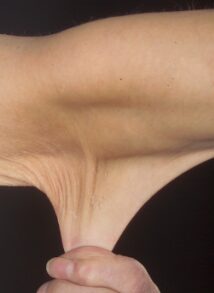
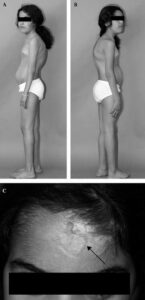
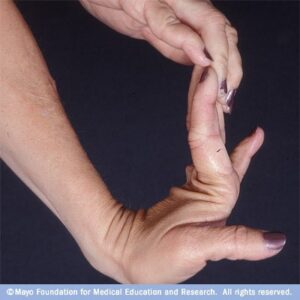
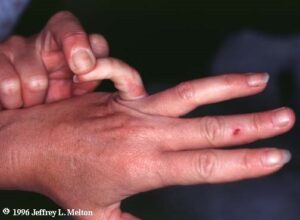
مفاصل بیش از حد انعطافپذیر هستند و امکان دررفتگی اسکلتی بخصوص در ناحیه باسن و پاها در هنگام تولد یا در مرحله رشد کودک با کمترین ضربه فیزیکی وجود دارد. مبتلایان به سندرم EDS علائمی همچون: قفسه سینه پیچ خورده، شروع زودرس استئوآرتریت پیشرفته، دژنراتیو مفاصل، ناهنجاری گردن و انگشتان دست، پارهگی تاندن یا عضلات و تغییر شکل در ستون فقرات از جمله اسکولیوز ( انحنای ستون فقرات )، قوز سینهای و درد شدید عضلانی و مفصلی را نشان میدهند.
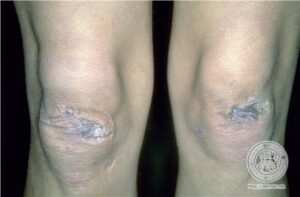
علائم پوستی :
مبتلایان به سندرم EDS دارای پوست بسیار شکننده که با آسانی با کمترین تحریک کبود میشود. این افراد در هنگام استفاده از سرویسهای بهداشتی منجمله استحمام بایستی به آهستگی پوست خود را شستشو دهند چرا که با کمترین فشار، پوست آنها دچار پارگی مویرگی شده و کبود میشود.
قلب و عروق :
افراد مبتلا به سندرم EDS دچار پارگی شریانها شده که بیماری دریچه قلبی را به همراه خواهد داشت. همچنین پارگی آئورت نیز وجود دارد که میتواند تهدید کننده حیات باشد.
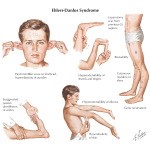
ژنتیک مولکولی سندرم اهلرز- دانلوس :
سندرم EDS هم تحت تاثیر توارث مادری یا پدری بوده و هم تحت تاثیر اپیژنتیک میباشد. اما آنچه در سندرم EDS مدنظر است جهش در ژنهای COL5A2،COL5A1،COL3A1،COL1A2،COL1A1 و TNXB و همچنین جهش در آنزیمهای DSE،B4GALT7،POLD1،ADAMTS2،D4ST1 و CHST14 میباشد. جهش در این ژنها معمولا ساختار، تولید و یا پردازش کلاژن را تغییر میدهد. نقص در کلاژن میتواند قدرت بافت همبند در پوست، استخوانها، رگهای خونی و اندامها را تضعیف کند و در نتیجه دچار اختلال کند.
تشخیص سندرم EDS :
تشخیص را میتوان با ارزیابی تاریخچه پزشکی و مشاهدات بالینی انجام داد. در تشخیص سندرم EDS معیار Brighton بطور گسترده برای ارزیابی درجه تحرک مفاصل استفاده میشود. قطعیترین روش تشخیص، انجام آزمایش DNA و مطالعات بیوشیمیایی میباشد. آزمونهای تشخیصی ژنتیکی عبارتند از: آزمایش جهش ژن کلاژن، نمونهبرداری از پوست، اکوکاردیوگرام و آزمایش فعالیت اکسیداز. با این حال ارزیابی بالینی توسط متخصص ژنتیک امری ضروری است. این سندرم در حیوانات هم وجود دارد و میتواند حیوانات را با علائم مشابه انسانی درگیر سازد.
مسیرهای درمانی :
هیچ درمان قطعی برای سندرم EDS وجود ندارد اما میتوان با حمایت بیماران از نظر جراحی و نظارت نزدیک گوارش و درمان فیزیکی رنج بیماران را تخفیف داد. افرادی که از EDS رنج میبرند اغلب با اضطراب و اندیشه بیهوده راجع سلامتی خود دچار افسردگی و خستگی مزمن میشوند. از آنجا که EDS ناتوانی نامرئی در نظر گرفته میشود، بسیاری از افراد مبتلا مشکلات روانی و اجتماعی ناخوشایند از خود نشان می دهند.
References:
- Wenstrup et al.،J (2001). The Ehlers–Danlos Syndromes: Management of Genetic Syndromes. pp. 131–149.
- Lind J، Wallenburg HC (April 2002). “Pregnancy and the Ehlers–Danlos syndrome: a retrospective study in a Dutch population”. Acta Obstetricia et Gynecologica Scandinavica 81 (4): 293–300.
- Guilleminault C، Primeau M، Chiu HY، Yuen KM، Leger D، Metlaine A (2013). “Sleep-disordered breathing in Ehlers-Danlos syndrome: a genetic model of OSA”. Chest 144 (5): 1503–11.
- Rombaut L، Malfait F، De Wandele I، Cools A، Thijs Y، De Paepe A، Calders P (July 2011). “Medication، Surgery، and Physiotherapy Among Patients With the Hypermobility Type of Ehlers-Danlos Syndrome”. Archives of physical medicine and rehabilitation 92 (7): 1106–1112.
- Woerdeman LA، Ritt MJ، Meijer B، Maas M، (2000). “Wrist problems in patients with Ehlers-Danlos syndrome”. European Journal of Plastic Surgery 23 (4): 208–210.
- Callewaert B، Malfait F، Loeys B، De Paepe A (March 2008). “Ehlers-Danlos syndromes and Marfan syndrome”. Best practice & research. Clinical Rheumatology 22 (1): 165–189.
- Byers PH، Murray ML (2012). “Heritable collagen disorders: the paradigm of Ehlers-Danlos syndrome”. Journal of Investigative Dermatology 15 (132): E6–11.
- Beighton P، De Paepe A، Steinmann B، Tsipouras P، Wenstrup RJ (1998). “Ehlers–Danlos syndromes: revised nosology، Villefranche، Ehlers–Danlos National Foundation (USA) and Ehlers–Danlos Support Group (UK)”. American Journal of Medical Genetics 77 (1): 31–7.
- Levy، Howard (2004). “The Ehlers Danlos Syndrome، Hypermobility Type.” University of Washington: NIH.
- Erçöçen AR، Yenidünya MO، Yilmaz S، Ozbek MR (1997). “Dynamic swan neck deformity in a patient with Ehlers-Danlos syndrome”. J Hand Surg Br 22 (1): 128–30.
- Milhorat TH، Bolognese PA، Nishikawa M، McDonnell NB، Francomano CA (December 2007). “Syndrome of occipitoatlantoaxial hypermobility، cranial settling، and chiari malformation type I in patients with hereditary disorders of connective tissue”. Journal of Neurosurgery. Spine 7 (6): 601–9.
- Vigorita، Vincent J; Mintz، Douglas & Ghelman، Bernard (2008). Orthopaedic pathology (2nd ed. ed.). Philadelphia: Lippincott Williams and Wilkins. pp. 5–6.
- Halper et al. “Glycan profiling of a defect in decorin glycosylation in equine systemic proteoglycan accumulation، a potential model of progeroid form of Ehlers–Danlos syndrome”. Department of Pathology، College of Veterinary Medicine، The University of Georgia، Athens، GA 2010.
- com. Retrieved 2014-02-27.
https://www.ehlers-danlos.com/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام
سلام بنده وفرزندانم دارای بیماری اهلرز دانلوس هستیم، جهت تشکیل انجمن حمایتی خواهان جمع اوری بیماران دیگر هستم لطفا با شماره زیر پیام بدید 09179113924