سندرم شواخمان- دیاموند
الهام علیزاده میلانی و شاهین اسعدی دانشجویان ژنتیک مولکولی
دکتر علی نظیرزاده متخصص ژنتیک، استادیار دانشگاه


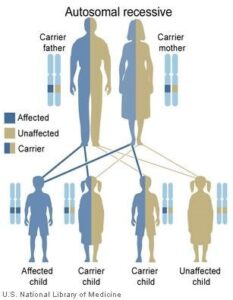
سندرم شواخمن دیاموند (SDS) نوعی بیماری مادرزادی ارثی است که با نارسایی بخش برونریز پانکراس، اختلال کارکرد مغز استخوان، ناهنجاری اسکلتی و کوتاهی قد همراه است. پس از بیماری فیبروز کیستیک شایعترین علت نارسایی بخش پانکراس کودکان است و علائم اولیه این سندرم مشابه بیماری فیبروز کیستیک است. این سندرم یک بیماری اتوزومال است که به صورت مغلوب به ارث میرسد، یعنی اگر یکی از والدین مبتلا به این بیماری باشد به نسبت مساوی در فرزندان دختر و پسر احتمال بروز وجود دارد. ژن جهشیافته در این بیماری SBDS است که روی بازوی بلند کروموزوم شماره 7 به صورت 7q1122 قرار دارد.

این بیماری برای اولین بار در سال 1964توسط Boldian Sheldon & lightwood به عنوان نهاد بالینی شناسایی شد و در همان سال پروسه ژنتیکی آن توسط khaw & shwachman diamond کشف شد.
بطور کلی SBDS در تشکیل ریبوزوم نقش دارد و جهش درSBDS و پروتئین آن منجر به نقص در مونتاژ ریبوروم بالغ میشود. همچنین SBDS در متابولیسم RNA شرکت داشته و با کمک این آنزیم در سلولهای استرومای سالم باعث تشکیل زیرواحد 80s ریبوزومی میشود. اگر جهش در ژن SBDS رخ دهد، ژن elF6 در نبود SBDS مانع اتصال دو زیرواحد 40s,60s میشود و در نتیجه عملکرد ریبوزوم دچار اختلال میگردد. SBDS در افزایش آپوپتوزیس در سلولهای اریتروئید و ایجاد اریتروپوزیس نرمال نقش ایفا میکند.

علائم بیماری
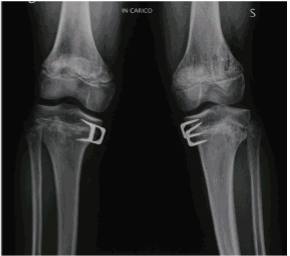
عقب ماندگی جسمی از علائم این بیماری است که با اختلال در رشد استخوان ران و زانو همراه است و فرد دچار بیماری استئوپینا شده یعنی استخوان کمتراکم و شکننده میشود. نارسایی بخش برونریز پانکراس یکی دیگر از نشانههای سندرم شواخمن است که نحوه عملکرد لوزالمعده را مختل میکند. همانطور که میدانیم آنزیمهایی برای هضم غذا از پانکراس ترشح میشود که در اثر کمبود سلولهای Acinar که تحت تأثیر آنزیمهای گوارشی هستند مواد غذایی به خوبی هضم نشده و مواد مغذی به بدن بیمار جذب نمیشود.
یکی دیگر از علائم این بیماری ناهنجاری خونی است که در این بیماران پلاکت خون کم شده و فرایند انعقاد خون را مشکل میسازد. نوتروفیل به میزان کم تولید شده و یا حتی تولید نمیشود و این افراد را مستعد عفونت میسازد. این بیماران مستعد نوتروپنی، ترومبوسیتوپنی، آنمی آپلاستیک نیز هستند.

اثر شیمیوتاکسی در لوکوسیت سندرم شواخمن
همانطور که گفته شد جهش در تک ژن SBDS باعث SDS فرد میشود. آنالیز رفتار جهشی بنیادی و پاسخ کمیوتاکسی، لوکوسیت چند هستهای (PMNs) در بیماران SDS نشان داد که پاسخ SDS PMNs در عدم و حضور یک ماده کمیوتاکسی طبیعی است ولی پاسخ در افزایش و کاهش ماده محرک fmlp در یک رنج خاص نیز نرمال است و این بیماران که مستعد عفونتاند میتوانند در برابر عفونت پاسخهای التهابی طبیعی بدهند. گیرنده این ماده محرک در سراسر غشای پلاسمایی برای کنترل لوکوسیتها، حتی در بیماران وجود دارد اما نقص در این گیرنده و افزایش بیش از حد ماده محرک، میزان و زمان پاسخگویی التهابی در بیماران SDS مشاهده میشود.
مسیر مولکولی سندرم شواخمن دیاموند
SBDS در چندین مرحله بیولوژیک نقش دارد که شامل بلوغ ریبوزوم، تشکیل دوک تقسیم و حرکت خودبخودی سلولها میباشد، اما نقش منفی آن در بیماران SDS است که بیثباتی در دوک تقسیم میتوزی، پاسخدهی سریع سلول در استرسها و مهاجرت مونوسیت Rac2 و کاهش پتانسیل غشاء میتوکندری و افزایش آپوپتوزیس است. پروماتور SBDS و ETF6 بر روی زیرواحد 60s ریبوزوم، برای پیوستن به زیرواحد کوچکتر و برای تشکیل و عملکرد ریبوزوم 80s، باید وجود داشته باشد.

در فنوتیپ یک مخمر به علت کمبود sdo1 که از ارتولوگ SBDS،کندی رشد را نشان میدهد حاصل موتاژن در Tif6 است که باعث اختلال در اتصال Tif6 به زیرواحد ریبوزوم 60s میشود. معادلTif6 در پستانداران همان EIF6 میباشد. آزاد شدنEIF6 از زیرواحد ریبوزوم s60 توسط EFL1، SBDS و GTP صورت میگیرد. شروع جهش SBSD پاتوژنز، ELF6 آزاد شده را تخریب میکند. SBSD، محرک فعالیت GTPass در EFL1 میباشد که متعاقباً اتصال GTP به EFL1 موجب پایداری SBDS میشود.
دو سندرم دیگر شامل آنمی دیاموند- بلاکفن و دیسکیراتوزیس کانجینیتا مشابه سندرم شواخن دیاموند هستند که در آنمی آپلاستیک، نوتروپنی، میلودیسپلازی و نارسایی مغز استخوان مشترکاند. فرق کمخونی آپلاستیک با MDS در این است که در MDS سلولهای خونی نابالغ یا غیرعادی تولید میشوند ولی در آپلاستیک با کاهش شدید تولید سلولهای خونی مواجه هستیم.


راههای درمانی
نارسایی بخش برونریز پانکراس را میتوان با مکملهای آنزیمی پانکراسی درمان نمود. ترمیم ناهنجاری اسکلتی نیاز به عمل جراحی دارد. نوتروپنی را میتوان با فاکتور محرک گرانولوسیتی درمان کرد. پیوند مغز استخوان را میتوان در صورت نارسایی پیشرونده مغز استخوان انجام داد. در آینده نزدیک میتوان از ژن درمانی برای درمان این بیماری استفاده کرد.
References:
1. Orkin, Stuart H.; Nathan, David G.; Ginsburg, David; A. Thomas Look (2009). Nathan and Oski’s hematology of infancy and childhood
- Shammas C, Menne TF, Hilcenko C, Michell SR, Goyenechea B, Boocock GR, Durie PR, Rommens JM, Warren AJ (2005). “Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman–Diamond Syndrome.”. J Biol Chem. 280 (19): 19221–9. doi:1074/jbc.M414656200. PMID15701631.
- Austin KM, Leary RJ, Shimamura A (2005). “The Shwachman–Diamond SBDS protein localizes to the nucleolus.”. Blood 106 (4): 1253–8. doi:1182/blood-2005-02-0807. PMC1895203. PMID15860664.
- Makitie O, Ellis L, Durie PR, Morrison JA, Sochett EB, Rommens JM, Cole WG (2004). “Skeletal phenotype in patients with Shwachman–Diamond syndrome and mutations in SBDS.”. Clin Genet 65 (2): 101–12. doi:1111/j.0009-9163.2004.00198.x. PMID14984468.
- Goobie, S; Popovic, M; Morrison, J; Ellis, L; Ginzberg, H; Boocock, GR; Ehtesham, N; Bétard, C; Brewer, CG; Roslin, NM; Hudson, TJ; Morgan, K; Fujiwara, TM; Durie, PR; Rommens, JM (April 2001). “Shwachman-Diamond syndrome with exocrine pancreatic dysfunction and bone marrow failure maps to the centromeric region of chromosome 7.”. American Journal of Human Genetics 68 (4): 1048–54. doi:1086/319505. PMC1275624. PMID11254457.
- Popovic, M; Goobie, S; Morrison, J; Ellis, L; Ehtesham, N; Richards, N; Boocock, G; Durie, PR; Rommens, JM (April 2002). “Fine mapping of the locus for Shwachman-Diamond syndrome at 7q11, identification of shared disease haplotypes, and exclusion of TPST1 as a candidate gene.”. European journal of human genetics : EJHG 10 (4): 250–8. doi:1038/sj.ejhg.5200798. PMID12032733.
- Disorders, National Organization for Rare (2003). NORD guide to rare disorders. Lippincott Williams & Wilkins. pp. 417–. ISBN978-0-7817-3063-1. Retrieved 8 August 2011.
- Hassan, Fauziya; Byersdorfer, Craig; Nasr, Samya (1 January 2010). “Severe Shwachman-Diamond syndrome and associated CF carrier mutations”. Respiratory Medicine CME 3 (1): 18–19. doi:1016/j.rmedc.2009.02.001.
- Bodian M, Sheldon W, Lightwood R (1964). “Congenital hypoplasia of the exocrine pancreas.”. Acta Paediatr 53: 282–93. doi:1111/j.1651-2227.1964.tb07237.x. PMID14158482.
- Shwachman H, Diamond LK, Oski FA, Khaw KT (1964). “The syndrome of pancreatic insufficiency and bone marrow dysfunction.”. J Pediatr 65: 645–63. doi:1016/S0022-3476(64)80150-5. PMID14221166.
- Goobie S, Popovic M, Morrison J, Ellis L, Ginzberg H, Boocock GR, Ehtesham N, Betard C, Brewer CG, Roslin NM, Hudson TJ, Morgon K, Fujiwara TM, Durie PR, Rommens JM (2001). “Shwachman–Diamond syndrome with exocrine pancreatic dysfunction and bone marrow failure maps to the centromeric region of chromosome 7.”. Am J Hum Genet 68 (4): 1048–54. doi:1086/319505. PMC1275624. PMID11254457.
- Popovic M, Goobie S, Morrison J, Ellis L, Ehtesham N, Richards N, Boocock G, Durie PR, Rommens JM (2002). “Fine mapping of the locus for Shwachman–Diamond syndrome at 7q11,
- identification of shared disease haplotypes, and exclusion of TPST1 as a candidate gene.”. Eur J Hum Genet 10 (4): 250–8. doi:1038/sj.ejhg.5200798. PMID12032733.
سندرم اهلرز- دانلوس
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام