
اصول و کاربرد روش Tandem MS در پژوهش و آزمایشگاه بالینی
محمدهادی نعمتالهی
دانشجوی دکتری بیوشیمی بالینی
دکترغلامرضا اسدیکرم
استاد گروه بیوشیمی بالینی دانشکده پزشکی

از آنجائیکه چند سالی است که اسپکترومتری جرمی به عنوان یک روش مطمئن و قدرتمند به آزمایشگاههای رفرانس وارد شده است و انتظار میرود حیطه عملکردی آن در آزمایشگاههای تشخیصی روز به روز گسترش یابد بر آن شدیم تا برای آشنایی خوانندگان فرهیخته ماهنامه اخبار آزمایشگاهی با این روش متن زیر را گردآوری و تقدیم نمائیم و امیدواریم که مورد استفاده همکاران ارجمند قرار گیرد.
1- تاریخچه ابداع طیف سنجی جرمی
تاریخچه روش اسپکترومتری جرمی به سال 1898 به آزمایشات wien برمیگردد. او نشان داد اشعههای کانالی میتوانند توسط عبور از بین میدانهای مغناطیسی و الکتریکی موازی منحرف شوند. با این وجود ابداع این روش توسط J.J.Thomsone صورت گرفت. تامسون آنالیز اشعههای کاتدی را مطالعه کرد. در دهه1960 طیف سنج های جرمی پشت سرهم[1] ابداع شدند که ارزش بالایی در زمینه بررسی ساختار و شناخت دقیق و صحیح مواد نقش مهمی داشتند.
در دهه 1980 با ایجاد روشهای یونیزه کننده ملایمتر طیف کاربرد اسپکترومتری جرمی افزایش چشمگیری یافت. ابداع روشهای اخیر باعث شد کاربرد اسپکترومتری جرمی در زمینه بیولوژی و علوم گسترش یابد و در سال 2002 John fehn و koichi Tanaka به دلیل نشان دادن اینکه پروتئینها با وزن مولکولی بالا را نیز میتوان با استفاده از روش یونیزاسیون دفع لیزر یونیزه کرد، جایزه نوبل دریافت کردند(1).

تامسون
اولین طیف سنج جرمی در اوائل قرن20 توسط تامسون طرحریزی شد. در این دستگاه پرتوی از یونهای مثبت در لوله تخلیه ایجاد میگردد و از طریق یک مسیر باریک به صورت موازی خارج میگردد. سپس وارد یک جداکننده جرمی میگردند. در اینجا دو میدان مغناطیسی و الکتریکی به طور همزمان اعمال میشود. بعد از منحرف شدن یونها توسط میدانها، بر روی یک پرده فلورسانس تصویرهای سهمی را شکل میدهند و به این دلیل این دستگاهها را دستگاههای سهمی نیز مینامند(2). تامسون در کتاب خود به نام پرتوهای الکتریکی که در سال 1913 به چاپ رسید، در مقدمه آورده است:
“من مطمئنم که در شیمی مسائلی وجود دارد که میتوان آنها را با این دستگاه خیلی آسانتر از دستگاههای دیگر حل نمود. این روش حساسیت بسیار خوبی دارد و حتی برای تجزیه مقادیر ناچیز به کار گرفته میشود و لزومی ندارد که نمونه خالص گردد(3).”
2- اصول کلی دستگاه
اولین سوالی که به ذهن میرسد این است که اسپکترومتری جرمی چیست. اصول پایهای اسپکترومتر جرمی (MS) تولید یونهایی از ترکیبات آلی یا غیر آلی با روشهای مناسب میباشد که برای جدا کردن این یونها از نسبت جرم به بار (m/z) آنها استفاده میشود. در این روش، آنالیت ممکن است به روشهای متفاوت که شرح داده میشود، یونیزه گردد. جداسازی یونها نیز تحت تأثیر میدانهای الکتریکی یا مغناطیسی استاتیک و یا پویا قرار میگیرد(4). طیف سنج جرمی بر اساس درصد فراوانی یونها نسبت به m/z ترسیم میشود و نه جرم واقعی آن یون(5).

اجزاء تشکیل دهنده طیف سنج جرمی

نمونهای از پیکهای حاصل از طیف سنجی جرمی
2-2 اجزاء دستگاه
همه طیفسنجها از بخشهای اصلی زیر تشکیل شدهاند:
1- سيستم ورودي نمونه (sample inlet)
2- منبع توليد يون (Ion source)
3- دستگاه تجزيه كننده يون (Ion Analyzer System )
4- آشكار ساز يوني (Ion detector)
5 – سيستم ثبت طيف (Spectrum recording system)
6 – محفظه خلاء و پمپ خلاء (Vacaum chamber & pump System)
در طیف سنج جرمی، یون باید فاصله منبع یونیزاسیون تا آشکارساز را بدون هیچ گونه برخوردی با یونها و مولکولهای دیگر طی کند، عملاً چنین حالتی را با ایجاد فشاری کمتر از torr 10 به دست میآورند. نمونهها در منبع یونی، یونیزه شده سپس به وسیله آنالیزور جرمی بر اساس m/z از هم جدا میشوند و توسط آشکارساز شناسایی و شمارش میشوند.
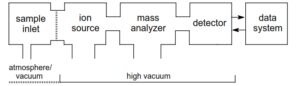
شکل کلی اجزا دستگاه طیف سنج جرمی
روشهای متفاوتی برای یونیزاسیون وجود دارد. به طور کلی روشهای یونیزاسیون به 2 روش یونیزاسیون soft (نرم) و Hard(سخت) تقسیمبندی میشوند و هر کدام کاربرد خاصی دارند. در یونیزاسیون سخت یونیزاسیون توﺳﻂ ﭘﺮﺗﻮيی از اﻟﮑﺘﺮونﻫﺎي ﭘﺮ اﻧﺮژي رخ میدهد ﺑﻪ دﻟﯿﻞ اﻧﺮژي زﯾﺎد ﻣﻮﺟﻮد در اﯾﻦ ﻓﺮآﯾﻨﺪ، ﯾﻮن ﻣﻮﻟﮑﻮل (Molecular Ion) ﺗﻮﻟﯿﺪ ﺷﺪه میتواند ﺑﻪ ﯾﻮن ﻣﻮﻟﮑﻮلﻫﺎي ﮐﻮﭼﮑﺘﺮ ﺗﺒﺪﯾﻞ ﺷﺪه ﯾﺎ دﭼﺎر ﻧﻮآراﯾﯽ ﺷﻮد ولی در یونیزاسیون نرم چون ﻧﻤﻮﻧﻪ ﺑﻪ ﺟﺎي ﭘﺮﺗﻮﻫﺎي ﭘﺮاﻧﺮژي ﺑﺎ ﯾﻮنﻫﺎ (ﮐﻪ ﻣﺴﻠﻤﺎ اﻧﺮژي ﺑﺮﺧﻮردي ﮐﻤﺘﺮي دارﻧﺪ) ﺑﺮﺧﻮرد ﻣﯽﮐﻨﺪ، ﯾﻮﻧﯿﺰاﺳﯿﻮن ﻣﻼﯾﻢﺗﺮ ﺧﻮاﻫﺪ ﺑﻮد.
ﻟﺬا ﺑﺮﺧﻼف روش ﺑﺮﺧﻮرد اﻟﮑﺘﺮون (EI)، اﺟﺰاي ﺳﺎﺧﺘﺎري ﻧﻤﻮﻧﻪ اﺻﻠﯽ دﭼﺎر ﺷﮑﺴﺖ ﺷﺪﯾﺪي ﻧﻤﯽﺷﻮﻧﺪ. برای مثال وجود یون مولکولی در طیف جرمی به ویژه اگر وزن جرمی به درستی اندازهگیری شده باشد ابزار بسیار ارزشمندی درباره شناسایی ترکیبات ناشناخته ارائه میدهد. برای دست یافتن به این هدف تکنیک یونیزاسیون نرم نیاز است تا فرآیند قطعه قطعه سازی در کمترین حد خود باشد. مثالهایی از تکنیک یونیزاسیون نرم یونیزاسیون شیمیایی، یونیزاسیون میدانی (FI) و روش الکترواسپری میباشد(8).
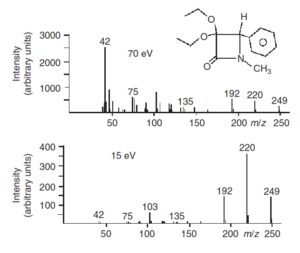 مقایسه یونیزاسیون نرم با سخت
مقایسه یونیزاسیون نرم با سخت
4-2 انواع روشهای یونیزاسیون
به طور کلی روشهای یونیزاسیون به روشهای زیر تقسیم بندی میشوند:
1-4-2 یونیزاسیون الکترونی (Electron Impact Ionization, EI)
هنگامی که یک الکترون (بار منفی) در یک میدان الکتریکی قرار گرفت، این الکترون انرژی جنبشی میگیرد. پس از تشدید در یک میدان 70 ولتی، الکترون انرژی حدود eV 70 (الکترون ولت) خواهد گرفت. این الکترون پرانرژی قادر است با مولکولهای خنثی و بدون بار برهمکنش دهد. نتیجه این پدیده خارج شدن الکترون از بالاترین تراز الکترونی مولکول خنثی و تشکیل یون از مولکول خنثی است.
![]()
یون ایجاد شده رادیکال کاتیون است. این پدیده در ابتدا برخورد الکترون (electron impact) نامیده شد. ﺑﻪ دﻟﯿﻞ اﻧﺮژي زﯾﺎد ﻣﻮﺟﻮد در اﯾﻦ ﻓﺮاﯾﻨﺪ، ﯾﻮن ﻣﻮﻟﮑﻮل (Molecular Ion) ﺗﻮﻟﯿﺪ ﺷﺪه میتواند ﺑﻪ ﯾﻮن ﻣﻮﻟﮑﻮلﻫﺎيﮐﻮﭼﮑﺘﺮ ﺗﺒﺪﯾﻞ ﺷﺪه ﯾﺎ دﭼﺎر ﻧﻮآراﯾﯽ ﺷﻮد که این یونها شکسته و به یونهای با جرم کمتر ( A و B و….) تبدیل میشوند. بیشتر یونهای ایجاد شده بوسیله روش یونیزاسیون الکتریکی تک بار هستند(10).
2-4-2 یونیزاسیون شیمیایی (CI, Chemical Ionization)
![]()
این روش به عنوان یک تکنیک نسبتاً جدید در طیف سنجی جرمی به کار میرود تا اطلاعاتی را که به وسیله بمباران الکترونی حاصل نمیشود به دست آوریم. یکی از معایب اصلی اطلاعات حاصله از بمباران الکترونی این است که پیک مولکولی ترکیبات خیلی ضعیف میباشد. اگر فراوانی یون مولکولی در روش بمباران الکترونی به اندازه 1 تا 2 درصد باشد، با این روش فراوانی نسبی آن به 10 تا 100 برابر میرسد. در واقع در حین بمباران الکترونی، مولکولها انرژی بیشتری از انرژی لازم برای یونیزاسیون دریافت کرده و در نتیجه یون مولکول با شکست پیوندها همراه میباشد که این امر باعث کاهش غلظت یون مولکول میگردد(2, 10).
![]()

یونیزاسیون شیمیایی ماده M توسط گاز متان
اما پدیده یونیزاسیون شیمیایی با انتقال انرژی کمتری همراه میباشد که در نتیجه عمل جزء به جزء شدن بسیار کاهش مییابد. در این روش یک گاز واکنش دهنده مثل متان یا ایزوبوتان توسط بمباران الکترونی یونیزه میشود. چنین مولکولهایی به نمونه برخورد کرده و عمل یونیزاسیون و جزء به جزء شدن نمونه مورد نظر انجام میشود(10, 11). ﺑﻪ ﻃﻮر ﺧﻼﺻﻪ ﻣﯽﺗﻮان ﮔﻔﺖ، از آن ﺟﻬﺖ ﮐﻪ در روش ﯾﻮﻧﯿﺰاﺳﯿﻮن ﺷﯿﻤﯿﺎﯾﯽ، ﻧﻤﻮﻧﻪ ﺑﻪ ﺟﺎي ﭘﺮﺗﻮﻫﺎي ﭘﺮاﻧﺮژي ﺑﺎ ﯾﻮنﻫﺎ (ﮐﻪ ﻣﺴﻠﻤﺎً اﻧﺮژي ﺑﺮﺧﻮردي ﮐﻤﺘﺮي دارﻧﺪ) ﺑﺮﺧﻮرد ﻣﯽﮐﻨﺪ، ﯾﻮﻧﯿﺰاﺳﯿﻮن ﻣﻼﯾﻢﺗﺮ ﺧﻮاﻫﺪ ﺑﻮد و بدین جهت آﻧﺎﻟﯿﺰﻫﺎﯾﯽ ﺟﻬﺖ ﺷﻨﺎﺳﺎﯾﯽ ﮐﯿﻔﯽ و ﺳﺎﺧﺘﺎري ﻧﻤﻮﻧﻪ ﻣﻌﻤﻮﻻً ﺑﺮ ﭘﺎﯾﻪ اﯾﻦ روش ﯾﻮﻧﯿﺰاﺳﯿﻮن ﺻﻮرت ﻣﯽﭘﺬﯾﺮد.
3-4-2 یونیزاسیون میدانی (Field Ionization)
مکانیسم آن به این صورت است که یک میدان الکتریکی با قدرت زیاد در حدود ( volt/cm 107 – 108) توسط یک تیغه باریک و یک سیم نازک که دارای ولتاژ خیلی زیاد ( volt 7000 – 10000 ) است، به وجود میآید. این قدرت زیاد میدان الکتریکی موجب میشود الکترون از یک سد پتانسیل انرژی مولکولی عبور کرده و منجر به تشکیل یون مولکولی گردد(12, 13).
4-4-2 یونیزاسیون گرمایی (Thermal Ionization)
اساس کار منبع یونیزاسیون گرمایی براین مبنا استوار است که وقتی اتمها یا مولکولهای با پتانسیل یونیزاسیون (ev) I بر روی یک سطح فلزی داغ با درجه حرارتی معادل (K) Tو تابع کار ev)w) قرار گرفته و گرم شوند، احتمال دارد که علاوه بر ذرات تبخیر شده یونهایی نیز به وجود آید(14).
5-4-2 یونیزاسیون به روش بمباران اتمی (Fast atom bombardment)
در اکثر طیف سنج های جرمی که دارای روشهای مختلف یونیزاسیون میباشند، نمونهها قبل از اینکه وارد اتاقک یونیزاسیون شوند باید به صورت بخار درآیند. حال با توجه به اینکه بعضی ترکیبات از نظر حرارتی پایدار نیستند این مسئله مهمترین محدودیت برای یونیزه کردن مواد در طیف سنجی جرمی به حساب میآید و عمدتاً به همین دلیل تعداد زیادی از ترکیبات مهم بیولوژیکی مثل کربوهیدراتها، پپتیدها، نوکلئوتیدها و غیره را با روش معمولی یونیزاسیون (بمباران الکترونی و یونیزاسیون شیمیایی) یونیزه نمیکنند، بلکه روش جدید و مهمی برای اینگونه ترکیبات ارائه شده که یونیزاسیون بمباران اتمی (FAB) نامیده میشود(2).
در FAB مواد مورد آزمایش با یک ماده شیمیایی غیر فرار به نام ماتریکس مخلوط میشود و تحت شرایط خلا با انرژی بسیار بالا پرتویی از اتمها بمباران میشوند. اتمها به طور معمول از گازهای خنثی مثل آرگون و زنون میباشند و ماتریکسهای رایج شامل گلیسرول، تیوگلیسرول، 3 ـ نیتروبنزیل الکل، سولفولان، دی اتانل آمین و تری اتانل آمین میباشد. FAB جز روش های یونیزاسیون soft طبقه بندی میگردد(15).
6-4-2 یونیزاسیون به روش الکترواسپری (Electrospray ionization)
یونیزاسیون به روش ESI اولین بار بوسیله DoIe و همکاران درسال 1968 گزارش شد. اما گروه Fenn در دانشگاه Yale برای اولین بار این روش یونیزاسیون را در طیفسنج جرمی استفاده کردند(16). در فرآیند یونیزاسیون به روش ESIیونهای موجود در محلول در فشار اتمسفری به فاز گازی منتقل میشوند و سپس از طریق یک سری منافذ وارد سیستم خلاء طیفسنج جرمی میشوند. در داخل منبع یونی ESI، یونها در شرایط فشار اتمسفری و با بهکارگیری جریان ملایم گاز نیتروژن از حلال آزاد میشوند. در مورد طیفسنجهای جرمی که منبع یونیزاسیون به کروماتوگرافی مایع متصل است (LC-MS) مخلوط نمونه در فاز مایع در کروماتوگرافی بر اساس اصول کروماتوگرافی از هم تفکیک شده و سپس اجزاء تفکیک شده به ترتیب از انتهای ستون کروماتوگرافی وارد منبع یونیزاسیون میشوند.
در این روش نمونه را در یک حلال قطبی فرار حل کرده و از درون یک لوله موئینه ضد زنگ (به قطر داخلی µm 75-100) با سرعت جریان 1 ml/min تا 1 µl/min پمپ میشود. در دهانه لوله مویینه ولتاژ نسبتاً بالایی ( KV4) اعمال میکنند، در نتیجه این عمل، قطرات بارداری از مخلوط نمونه و حلال از دهانه لوله به بیرون پاشیده (spray) میشوند. قطرات باردار به تدریج با وارد شدن به داخل محفظه منبع یونیزاسیون تبخیر شده و کوچک میشوند. در داخل منبع یونیزاسیون با استفاده از جریان ملایمی از گاز خشک نیتروژن در جهت حرکت یونها به سمت آنالیزور سبب تسریع تبخیر شدن قطرات و آزاد شدن یونهای نمونه میگردد. با تبخیر شدن حلال تراکم بارهای الکتریکی همنام افزایش یافته تا جایی که نیروی دافعه ناشی از بارهای همنام بر نیروی کشش سطحی قطره غلبه کرده و قطره شکسته شده و به قطرات کوچکتر تبدیل شود.
این فرآیند تا جایی که یونهای نمونه بطور کامل از قطره خارج شوند و حلال نیز به طور کامل تبخیر شود ادامه پیدا میکند. یونهای مثبت و منفی بسته به پتانسیل الکتریکی بکار رفته به دهانه لوله موئینه مهاجرت میکنند. اگر پتانسیل بکار رفته در انتهای لوله موئینه مثبت باشد یونهای منفی در دهانه لوله تجمع کرده در حالی که یونهای مثبت همراه با حلال به صورت قطرات بارداری وارد سیستم طیف سنج میشوند(17).
فرایند ESI به طور قابل ملاحظهای در دمای نسبتاً پایین (دمای اتاق) انجام میگیرد. بنابراین مولکولهای بسیار بزرگ، قطبی و مولکولهایی که از نظر حرارتی ناپایدارند را میتوان بدون تغییر ترکیب آنها با این روش یونیزه نمود. از این رو ESI را جزء روشهای یونیزاسیون ملایم (soft) طبقه بندی میکنند(18).
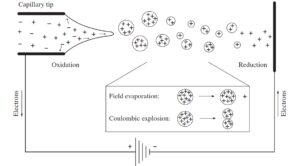
روش یونیزاسیون با الکترواسپری
7-4-2 یونیزاسیون به روش Matrix-assisted laser desorption ionization (MALDI)
برای اولین بار در سال 1988 توسط Hillenkamp و Karas برای آنالیز پروتئینها استفاده شد(19). از آن به بعد از MALDI بطور گستردهای در تحقیقات بیولوژی به ویژه مطالعه پروتئینها مورد استفاده قرار گرفت. همانند ESI، MALDI نیز قابلیت تبخیر و یونیزه نمودن مولکولهای بزرگ قطبی را دارد(20). با MALDI میتوان نمونههای با جرم Da 40000 آنالیز نمود. ایجاد یون در MALDI بر پایه بمباران نمونه با نور لیزر استوار است. از این نظر MALDI مانند یونیزاسیون به روش بمباران سریع اتمی (FAB) عمل میکند با این تفاوت که در FAB نمونه در ماتریکس گلیسرول حل میشود.
اما در MALDI نمونه به همراه ماتریکس جاذب نور لیزر کریستاله میشود. مکانیزم یونیزاسیون در MALDI بدین شرح است که ابتدا نمونه را با مقدار زیادی ماتریکس (Sinapinic acid و یا α-cyano-4-hydroxycinnamic acid) مخلوط میکنند.
مخلوط حاصل را روی صفحه فلزی میریزند و اجازه میدهند بطور کامل خشک شده و کریستاله شود. نقش ماتریکس، جذب نور لیزر در طول موج 337 نانومتر (لیزر نیتروژن) و کمک به پایداری نمونه است. تاباندن پالسهایی از نور لیزر (درحدود نانو ثانیه) منجر به جذب نور لیزر و گرم شدن سریع و تصعید مولکولهای ماتریکس به همراه نمونه میشود. هنگامی که ماتریکس به همراه نمونه وارد فاز گازی میشود، مقداری از انرژی مولکولهای برانگیخته شده ماتریکس به مولکولهای نمونه منتقل شده و سبب یونیزاسیون نمونه میشود(16).
البته علت اصلی یونیزاسیون مولکولهای نمونه هنوز بطور دقیق مشخص نیست. یونیزاسیون پپتیدها بیشتر به صورت مثبت و با جذب پروتون از مولکولهای ماتریکس صورت میگیرد. در واقع انرژی نور لیزر سبب برانگیخته شدن ماتریکس و جداشدن پروتون و انتقال آن به گروههای جاذب پروتون در ساختار پروتئین یا پپتید میگردد(17).
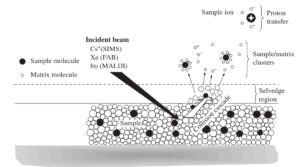
یونیزاسیون به روش MALDI
5-2 آنالیزورهای جرمی
قلب هر دستگاه طیفسنج جرمی را آنالیزور جرمی تشکیل میدهد. کار اصلی هر آنالیزور جرمی، جمع کردن یونهای با نسبت جرم به بار یکسان و متمرکز نمودن آنها به ترتیب یا بطور همزمان به داخل آشکارساز (در مورد طیفسنجهای معمولی) یا به داخل محفظه برخورد (درمورد طیفسنج جرمی متوالی) است. چون هر کدام از این آنالیزورهای جرمی بر پایه اصول متفاوتی عمل میکنند، هر یک مزایا و محدودیتهای خاص خود را دارند(21). در اینجا به انواع آنالیزورها اشاره میگردد:
1-5-2 آنالیزور مغناطیسی (magnetic sector analayzer)
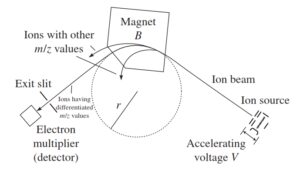
طیف سنجی مغناطیسی
دستگاههای طیف سنج جرمی در سالهای اخیر پیشرفتهای زیادی داشتهاند. به طور کلی هنگامیکه یون داخل یک میدان مغناطیسی تزریق میشود یک مسیر انحناداری را طی خواهد کرد که شعاع آن بستگی به جرم به بار و یا دقیقتر به mv دارد که vسرعت یون میباشد. در این میدان مغناطیسی هر چه نسبت جرم به بار بزرگتر باشد، شعاع انحنای مسیر نیز بزرگتر خواهد بود. لوله تجزیهگر دستگاه طوری طراحی شده است که شعاع انحنای ثابتی دارد. ذراتی که نسبت جرم به بار آنها بسیار بزرگ و یا بسیار کوچک باشند به کناره های لوله تجزیهگر برخورد کرده و به آشکارساز نمیرسد(2, 6).
2-5-2 آنالیزور تمرکز دوگانه (Double focusing analayzer)
همانطور که در آنالیزورهای جرمی مغناطیسی گفته شد فرآیند جداسازی یونها باید 2 کار را انجام دهد:
1- باید یونهایی که دارای جرم به بار متفاوت هستند را جدا سازد.
2- باید تمام یونهایی که دارای جرم به بار یکسانی هستند را تمرکز مسیر بخشد.
تجزیهکننده مغناطیسی هر دو کار مذکور را انجام میدهد ولی یک فاکتور دیگر وجود دارد که باعث پیچیدهتر کردن مسئله تمرکز مسیر میگردد و آن این است که همه یونهایی که دارای جرم به بار یکسانی هستند دارای سرعت یکسانی نیستند(10). یکی از تغییر دهندههای سرعت یونها، انرژی جنبشی است که یونها قبل از شتاب گرفتن دارا میباشند.

طیف سنجی تمرگز دوگانه
برای انجام تفکیک بهتر از طیف سنجهای جرمی تمرکز دوگانه استفاده میگردد. در چنین دستگاههایی پرتو یونها قبل یا بعد از ورود به میدان مغناطیسی از یک میدان الکتریکی عبور میکنند که در حضور میدان الکتریکی ذرات همگی با یک سرعت حرکت کرده و بنابراین قدرت تفکیک بهتر میگردد. برای بسیاری از کاربردها قدرت تفکیکی بیشتر از طیف سنجهای تمرکز دوگانه قدرت تفکیک تا 10 برابر افزایش مییابد(2).
3-5-2 آنالیزور جرمی چهار قطبی Quadrupole(Q)
آنالیزورجرمی Q از چهار میله موازی با مقطع هذلولی (هیپربولیک) ساخته شده است. هر یک از این میــــلهها طولی در حدود 2 /0 متر و قطری در حدود 6 میلی متر دارند. جفت لولههایی که بطور قطری مقابل یکدیگر قرارگرفته اند به ولتاژ dc (Direct current) مثبت و rf (radio frequency) متصل میشوند و جفت دیگر به ولتاژ dc منفی و ولتاژ rf که 180 درجه خارج از فاز است، متصل میشوند(16, 22). بنابراین یک میدان چهار قطبی در فضای بین لولهها ایجاد می شود.
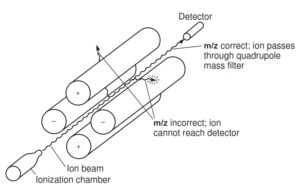
نحوه قرارگیری لوله های تشکیل دهنده و مسیرحرکت یون در Quadrupole
یونهایی که در منبع یونی ایجاد شدهاند، بطور الکترواستاتیکی در داخل این میدان تشدید میشوند. یونهای با نسبت جرم به بار متفاوت از طریق تغییر در ولتاژ جریان dc یا rf در فرکانس ثابت از هم جدا میگردند. تنها یونهای با نسبت جرم به بار مشخص در دامنه ولتاژ اعمال شده، مسیر حرکت پایدار داشته و قادرند به آشکارساز برسند(22). یونهای دیگر که مسیر حرکت ناپایدار دارند به میله ها برخورد کرده و از بین میروند. بنابراین Qدر هر زمان فقط اجازه عبور به یونهای با m/z مشخص میدهد(21).
4-5-2 آنالیزور جرمی تله یونی Ion trap) )
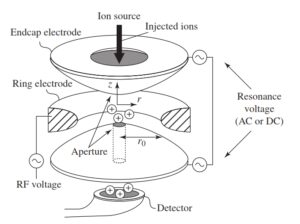
آنالیزور جرمی (Ion trap)
مکانیزم Ion trapهمانند Quadrupole است. از نظر مفهومی Ion trapهمان Quadrupole است که به دور خود پیچیده و تشکیل حلقه بستهای داده است. هر Ion trap از یک الکترود حلقوی و دو الکترود cupـend ساخته شده است. میدان درون تله (trap) به گونهایست که یونهایی که وارد آنالیزور میشوند، شروع به نوسان ثابتی میکنند و در آنالیزور به دام میافتند. سپس با تغییر ولتاژ rf بکار رفته در الکترود حلقوی امکان خروج یونهای خاصی فراهم میشود. برای اسکن نمودن یونها ولتاژ rf اعمال شده روی الکترود حلقوی را افزایش میدهند.
در همین لحظه ولتاژ rf کوچکی نیز به الکترود end cup وارد میکنند. بسامد حرکت نوسانی یون نیز افزایش مییابد. زمانی که بسامد تشدید (Resonant Frequency) یک یون به بسامد الکترود endcapمیرسد، حرکت نوسانی یون افزایش یافته و حرکت یون ناپایدار شده و در نهایت در طول محور الکترود endcap (trap) را ترک میکند. چون بسامد نوسان یون تابعی از جرم آن است، یونهای با نسبت جرم به بار (m/z) مختلف، تله (trap) را در ولتاژهای متفاوت و بنابراین در زمانهای متفاوتی ترک میکنند(16).
افزایش فرکانس جریان متناوب (rf) منجر به خروج یونها به ترتیب افزایش نسبت m/z از آنالیزور میشود(20). یونهای خارج شده از آنالیزور به سمت آشکار ساز هدایت میشوند. بر اساس فرکانس بکار رفته در هنگام خروج یون از تله یونی نسبت جرم به بار محاسبه میشود. تله یونی در مقایسه با آنالیزور TOF از حساسیت بیشتر و قدرت تفکیک و دامنه جرمی کمتری برخوردار است(21).
5-5-2 آنالیزور جرمی زمان پرواز ( Time Of Flight, TOF )
اندازهگیری نسبت جرم به بار در آنالیزور TOF براساس این اصل که اعمال پتانسیل شتابدهی (V) به یون با بار (Z)، انرژی جنبشی معادل zV به یون خواهد داد، انجام میگیرد(11). بنابراین یونهای با جرم متفاوت مسافت یکسان را در مدت زمان متفاوتی طی میکنند. نسبت جرم به بار بوسیله اندازهگیری زمانی که یون فاصله بین منبع یونی و آشکارساز (Fligh path) را طی میکند، بدست میآید.
TOF در مقایسه با سایر آنالیزورهای جرمی دارای قدرت تفکیک جرمی بهتر و دامنه جرمی بالاتری است. هر چه مسیر فرار (Flight Path) طولانیتر باشد، قدرت تفکیک بهتری حاصل میشود. در آنالیزورهای TOFتجاری، معمولاً مسیر فرار را چندین متر در نظر میگیرند. در برخی از آنالیزورهای جرمی TOF، یک آیینه منعکس کننده یون (Ion Reflector) در انتهای لوله فرار قرار میگیرد که سبب منعکس شدن یون به عقب و به داخل لوله فرار میگردد. از طریق این آیینه سبب افزایش طول لوله فرار میشود(23).
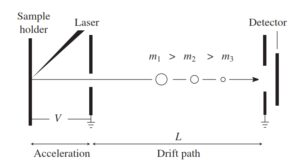
آنالیزور جرم TOF
6-5-2 آنالیزور جرمی Transform Fourier (FT )
آنالیزور جرمی FT ،( ICR ) Ion Cyclotron Resonance نیز نامیده میشود. در واقع FT نوعی Ion Trap متشکل از یک محفظه مکعبی شکل است که در یک میدان مغناطیسی قوی قرارگرفته است. این محفظه مکعبی شکل از سه دسته صفحه Trapping ،Transmitter وReciever که بطور دو به دو مقابل یکدیگر قرار گرفتهاند ساخته شده است. درک مکانیزم عمل FT در واقع از این واقعیت سرچشمه گرفته است که زمانی که یک یون در یک میدان مغناطیسی ثابت قرار میگیرد شروع به چرخیدن حول یک محور عمود بر جهت میدان میکند(18).
در مورد طیفسنج جرمی یونهایی که در منبع یونیزاسیون ایجاد شدهاند، زمانی که به داخل محفظه آنالیزور FT هدایت میشوند تحت تأثیر میدان مغناطیسی در جهت عمود بر میدان مغناطیسی اعمال شده شروع به چرخش کرده و در داخل محفظه به دام میافتند. بسامد حرکت دورانی یون با شدت میدان مغناطیسی نسبت مستقیم و با m/z یون نسبت عکس دارد.
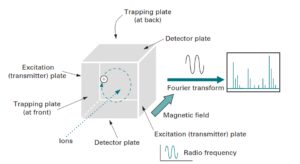
آنالیزور جرمی فوریه
7-5-2 آنالیزور جرمی اوربیترپ (orbitrap)
قدرت تفکیک متناسب با قدرت میدان مغناطیسی به کار رفته میباشد. بنابراین به یک آهنربای فوق هادی نیاز است که هزینه زیادی برای نگهداری چنین آهنرباهایی صرف میگردد. آنالیزور جرمی اوربیترپ هزینه معقولتر و مناسبتری دارد. یونها در اوربیترپ، جایی که یونها تحت نوسان هماهنگ هستند، در یک محور طولی از یک میدان الکتریکی به دام میافتند. مقادیر هر یون از روی فرکانس آن یون در یک روش غیر مخرب اندازهگیری شده و تبدیل به فوریه میگردد تا در نهایت طیف جرمی به دست آید. چنین دستگاههایی دارای قدرت تفکیک بسیار بالا( بیشتر از 150000) و حساسیتی در مقادیر در حد فمتومول میباشد(5).
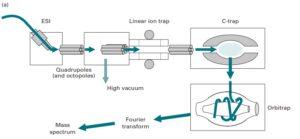
آنالیزور جرمی اوربیترپ با منبع ESI
6-2 آشکارساز (Detector)
یونهای با نسبت جرم به بار یکسان که توسط آنالیزور جرمی از سایر یونها جدا شدهاند، بصورت پرتوی به آشکارساز میرسند. آشکارساز تعداد یونهای با نسبت جرم به بار یکسان را شمارش میکند (تعداد یونها نشان دهنده فراوانی نسبی آنها است). روی محور افقی (xها) نسبت جرم به بار و روی محور عمودی (yها) شدت هر یون (فراوانی نسبی) نشان داده میشود. نوع آشکارساز را متناسب با نوع آنالیزور انتخاب میکنند. Electron multiplier و Microcannel دو آشکارسازی هستند که به طور گسترده در طیفسنجهای جرمی مورد استفاده قرار میگیرند(33).
تقویت کنندههای الکترون و مبدل دینود به عنوان یک آشکارساز در اکثر طیفسنجهای جرمی کاربرد دارد. در این نوع آشکارسازها یون به یک صفحه فلزی برخورد کرده و سبب ساطع شدن یک پرتوالکترونی و ایجاد یک جریان قابل اندازهگیری میشود(5).
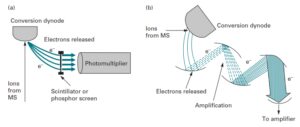
مبدل دینود و تقویت کننده الکترون
3-طیف سنجی متوالی و کاربرد بالینی آن
1-3 توالی یابی پپتیدها بوسیله طیف سنج جرمی متوالی (MS/MS)
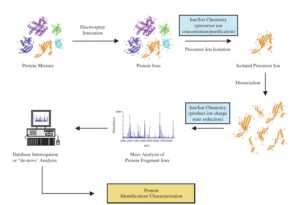
برای بدست آوردن اطلاعات ساختاری و توالی پپتیدی، از طیف سنج متوالی (MS2 و MS\MS) استفاده میکنند. در طیفسنج جرمی متوالی بطور معمول از دو آنالیزور جرمی که با یک محفظه برخورد (Cillision Cell) از هم جدا شدهاند و یا یک آنالیزور جرمی Ion trap استفاده میشود. در حالتی که از دو آنالیزور استفاده میشود در فاصله بین دو آنالیزور جرمی یک محفظه برخورد قرار میگیرد که در آن با بکارگیری یک گاز خنثی مانند هلیوم و یا آرگون طی فرایند Collision Induced Dissociation (CID) یون انتخابی را قطعه قطعه میکنند.
قطعه قطعه شدن در اثر برخورد مولکولهای گاز به مولکول یون رخ میدهد. آنالیزورهای جرمی مورد استفاده ممکن است از یک نوع یا از انواع مختلف (هیبرید) باشند. وقتی از یک نوع باشند میتوان به آنالیزورهای triple Quadupole اشاره کرد. برای آنالیز بوسیله MS/MS (طیف سنج جرمی متوالی) سه Quadrupole را با هم به شکل Triple Quadruple ترکیب میکنند. در اینجا اولین و سومین Quadrupole برای Scanning (تفکیک یونها براساس m/z) بکار میروند در حالی که Quadrupole میانی به عنوان محفظه برخورد عمل میکند. آنالیزور اول تنها اجازه عبور یونهای انتخابی توسط کاربر را فراهم میکند.
در حالی که آنالیزور سوم یونهای حاصل از قطعه قطعه شدن یون انتخابی در محفظه برخورد را، براساس نسبت جرم به بار از هم تفکیک کرده و طیف MS/MS را بوجود میآورد. در محفظه برخورد یونها طی فرایند CID و بمباران با گازهای خنثی نظیر هلیوم و آرگون قطعه قطعه میشوند(5).
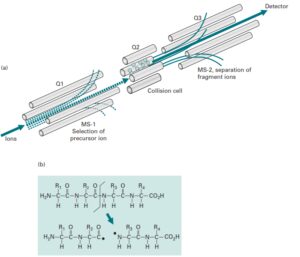
طیفسنج triple quarupole
طیفسنج متوالی میتواند از نوع هیبرید نیز باشد که ممکن است آنالیزور اول Quadrupole و آنالیز دوم TOF باشد.
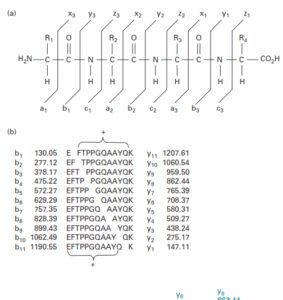
نمونه ای از نامگذاری یونهای شایع و تعیین توالی پروتئین
اولین آنالیزور جرمی امکان عبور همه یونهای نمونه را فراهم میکند در حالیکه آنالیزور ثانویه به گونهای تنظیم میشود تا تنها امکان عبور یونهای حاصل از بمباران یون انتخابی فراهم شود. آنالیزور دوم یونهای حاصل از شکست یون انتخابی را بر اساس نسبت جرم به بار از هم جدا کرده و آشکارسازی که در انتهای طیف سنج قرار دارد، تعداد یونهای متناظر هر قطعه را ثبت میکند. طیف بدست آمده جرم هر قطعه را مشخص مینماید.
در حالتی که از آنالیزور جرمی تله یونی (Ion tarp) استفاده شود، تمام یونها بجز یون مورد نظر با اعمال ولتاژ rf مناسب به الکترود حلقوی از تله یونی خارج میشوند. پس از به دام انداختن یون مورد نظر با بکارگیری گاز خنثی طی فرایند CID آنرا قطعه قطعه میکنند. سپس قطعات حاصله بر اساس نسبت m/z از هم تفکیک شده و طیف MS/MS را بوجود میآورند(21).
2-3 شناسایی پروتئینها بوسیله طیف سنج جرمی
توالییابی ژنوم برخی از موجودات زنده زمینههای تحقیقاتی بیولوژی مولکولی را متحول کرده است. واژه پروتئوم برای اولین بار در سال 1994 توسط ویلکینز برای توصیف مجموعه پروتئینهای بیان شده بوسیله یک ژنوم پیشنهاد شد(24). علم پروتئومیکس به مطالعه پروتئومها میپردازد. در حقیقت پروتئومیکس مطالعه بیان ژنها در سطح پروتئینها است. برای درک چگونگی کار سلولها مطالعه پروتئینها، نحوه عمل و چگونگی برقراری اثر متقابل با دیگر پروتئینها ضروری است. برخلاف طبیعت ثابت ژنوم که در همه سلولهای یک موجود یکسان است، پروتئوم دائماً تحت تحریکات محیطی و داخلی تغییر میکند(25).
تهیه فهرست تمام پروتئینهای کد شده توسط ژنوم یک موجود و تجزیه و تحلیل ساختاری و کنش این پروتئینها هدف اصلی پروتئومیکس است. توالییابی به روش ادمن برای مدت 25 سال یک روش استاندارد خوب برای توالییابی پروتئینها بود. اما پروتئین مورد مطالعه قبل از توالییابی بایستی خالصسازی و هموژنیزه شود و سپس طی چرخههایی دستخوش واکنشهای هضم ادمن قرار گرفته و توالی آن تعیین شود. محدودیت اصلی روش ادمن سرعت پایین آن و نیاز به پروتئین خالص است. درحال حاضر روشهای شناسایی پروتئینها بوسیله طیف سنج جرمی مبتنی بر دو استراتژی اصلی up ـ Bottom و Top-down هستند(26).
1-2-3 شناسایی پروتئینها به کمک روشهای مبتنی بر استراتژی Bottom-up
این روش شناسایی پروتئینها اولین بار با بکارگیری تکنیکهای ژل الکتروفورز یک بعدی و دو بعدی استفاده شد. در این جا لکه پروتئینی را از ژل خارج میکنند و سپس مورد هضم آنزیمی قرار میگیرد (بطور معمول از آنزیم تریپسین برای این کار استفاده می کنند). آنزیم تریپسین پیوند پیتیدی را از انتهای کربوکسیل اسیدهای آمینه لیزین و آرژینین برش میدهد. مخلوط پیتیدی حاصل از هضم آنزیمی بوسیله طیفسنج جرمی MALDI- TOF و یا ESI-MS بررسی میشود. جرم پیتیدهای حاصل از هضم، اثر انگشت جرم پپتیدی[2] (PMF) را به وجود میآورند.
در روش انگشتنگاری جرم پپتیدی، جرم پپتیدهای حاصل از هضم آنزیمی پروتئین ناشناخته با جرم تئوریکی پپتیدهای حاصل از هضم فرضی پروتئینهای موجود در بانک های اطلاعاتی مقایسه میشود(19).
بدست آوردن جرم دقیق پپتیدها برای شناسایی دقیق پروتئین یک ضرورت اجتناب ناپذیر است. بنابراین فاکتورهایی که سبب تغییر جرم پپتید میشوند سبب کاهش دقت جرمی نیز میشوند. یکی از این فاکتورها تغییرات پس از ترجمهای است که در ساختار پروتئینها رخ میدهد. اگر یک پروتئین ناشناخته به طور گستردهای تغییرات پس از ترجمه یافته باشد، پپتیدهای حاصل از آن با پپتیدهای حاصل از هضم پروتئین تغییر نیافته در بانک اطلاعاتی همخوانی نخواهد داشت.
PMF را نمیتوان بر روی یک مخلوط پروتئینی بکار برد چون هضم مخلوط پروتئینی ایجاد یک مخلوط پیچیده پپتیدی میکند که سبب افزایش پیچیدگی PMF میشود. بنابراین حتی اگر لکه پروتئنی جداشده از ژل دارای 2 تا 3 پروتئین باشد امکان شناسایی آن با PMF وجود نخواهد داشت(21).
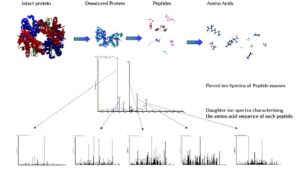
شناسایی پروتئین ها بوسیله انگشت نگاری جرم پپتدی (PMF)
2-2-3 شناسایی پروتئینها به وسیله روشهای مبتنی بر استراتژی Top-down
جرم پروتئینی به تنهایی برای شناسایی پروتئینی که از قبل شناسایی نشده است کافی نیست. تغییرات پس از ترجمهای که روی پروتئینها صورت میگیرد منجر به تفاوت جرم پیشبینی شده با جرم اندازه گیری شده میشود. اخیراً روشهایی مبتنی بر Top–down برای شناسایی پروتئینها توسعه یافتهاند که قادرند اطلاعاتی درباره ساختار اولیه پروتئین اصلی (intact) بدون نیاز به هضم آنزیمی فراهم آورند. در این روش، پروتئین اصلی را یونیزه میکنند سپس یونهای حاصل را در طیفسنج جرمی قطعه قطعه میکنند و طیف جرمی قطعات حاصل را بدست میآورند.
سپس با جستجو در بانکهای اطلاعاتی و با کمک توالی نشانمند (Sequence tag) پروتئین را شناسایی میکنند. مزیت اصلی روشهایTop–down انجام MS/MS بر روی پروتئین اصلی است که امکان شناسایی توالی کامل پروتئین و تغییرات پس از ترجمه بر روی پروتئین وجود دارد. مشکل اصلی روش Top-down یونیزه کردن پروتئینهای بزرگ است که با روشهای یونیزاسیون فعلی این کار بسیار دشوار است(21, 26).
شناسایی پروتئین ها به روش Top-down
3-3 شناسایی تغییرات پس از ترجمه (PTM ) بوسیله طیفسنج جرمی
درحقیقت تغییرات پس از ترجمه وقایع پردازشی شیمیایی هستند که خواص پروتئینها را پس از ترجمه تغییر میدهند. بنابراین تغییرات پس از ترجمه ساختار سوم و چهارم پروتئین را تعیین و فعالیت پروتئین را تنظیم میکنند(28). تغییرات پس از ترجمه به دو صورت روی پروتئینها اعمال میشوند، یا با برش پرتئولیتیک و یا با اضافه شدن گروههای خاصی به زنجیرههای جانبی اسیدهای آمینه در ساختار پروتئین. تغییرات پس از ترجمهای که بطور معمول در ساختمان پروتئین رخ میدهند شامل؛ فسفوریلاسیون، گلیکوزیلاسیون، استیلاسیون، متیلاسیون، سولفاته شدن، تشکیل پیوند دیسولفیدی، آمین زدائی و یوبیکوئیتینه شدن است. بسیاری از پروتئینهای یوکاریوتی پس از ترجمه دستخوش این تغییرات میشوند.
بسیاری از این تغییرات تنظیمی و قابل برگشت هستند. زمانی که پروتئینی در ژل شناسایی شد گام بعدی شناسایی تغییرات پس از ترجمهایست که ممکن است در آن وجود داشته باشد. البته گاهی امکان شناسایی برخی از تغییرات پس ترجمه مانند فسفوریلاسیون و گلیکوزیلاسیون در ژل وجود دارد. برای مثال، اضافه شدن یک گروه فسفات به پروتئین منجر به اضافه شدن دو بار منفی به بار خالص پروتئین میشود که سبب تغییر pH ایزوالکتریک پروتئین بدون تغییر قابل مشاهدهای در وزن مولکولی پروتئین میشود. بنابراین موقعیت پروتئین فسفوریله شده در ژل به طور افقی تغییر میکند. برخی از تغییرات پس از ترجمه حلالیت پروتئینها را تغییر میدهند.
اضافه شدن گروههای شیمیایی به پروتئینها منجر به تغییر جرم مولکولی پیتیدهای حاصل از هضم پروتئین میشود. از این خصوصیت میتوان برای شناسایی این تغییرات بوسیله طیف سنج جرمی استفاده کرد(21). پیشرفتهای اخیر در تکنیکهای طیفسنجی جرمی امکان شناسایی سریع پروتئینها را فراهم نموده است. تاکنون بیش از 200 نوع تغییر پس از ترجمه شناسایی شده است. تغییرات پس از ترجمه در تمام جنبههای آبشارهای سیگنالی نقش مرکزی دارند. بنابراین شناخت تغییرات پس از ترجمه پروتئینها در مسیرهای پیام رسانی امکان یافتن اصول پایهای تنظیم سیستمهای بیولوژیکی را فراهم خواهد کرد(21).
فسفوریلاسیون و دفسفوریلاسیون در باقیمانده اسیدهای آمینه سرین(S)، ترئونین(T)، تیروزین(Y) و هیستیدین(H) در ساختار پروتئینها بسیار شناخته شده و تغییرات قابل برگشتی هستند که فعالسازی و غیرفعالسازی فعالیت آنزیمها و تنظیم اثرات متقابل مولکولها در مسیرهای پیام رسانی را بر عهده دارند. چون بخش بسیار کوچکی از پروتئینها در محیط زنده فسفریله شدهاند، از این رو برای مطالعه تغییرات فسفوریلاسیون پروتئینها استفاده از بازدارندههای فسفاتازی ضروری است. استیلاسیون و داستیلاسیون در انتهای آمین و باقیمانده اسید آمینه لیزین به عنوان رقیبی برای فسفوریلاسیون است. یوبیکوئیتینه شدن و دیوبیکوئیتینه شدن نقش بسیار مهمی در نشانمند کردن پروتئینها برای تخریب بوسیله پروتئازوم دارد.
گلیکوزیلاسیون که شامل اتصال کووالانسی اولیگوساکاریدها به باقیمانده اسید آمینه آسپاژین (به اصطلاح N-liked) یا سرین ـ ترئونین (O-linked) است. طیفسنج جرمی روش عمومی برای آنالیز تغییرات پس از ترجمه است. اکثر تغییرات پس از ترجمه سبب افزایش یا کاهش جرم مولکولی مورد انتظار میشود(28). آنالیز تغییرات پس از ترجمه به مراتب دشوارتر از شناسایی پروتئین است. چون اولاً: به روشهای بسیار حساسی برای شناسایی آنها نیاز است. برای مثال چون تنها ٪10 ـ 5 سوبستراهای پروتئینکینازها فسفریله شدهاند ما نیاز به ابزارهایی برای شناسایی پروتئین تغییر یافته (فسفریله شده) در سطح بسیار پایین داریم. ثانیاً پیوند کوالانسی بین گروه شیمیایی ناشی از تغییر پس از ترجمه و زنجیره پلیپپتیدی ناپایدار است. ثالثاً تغییرات پس از ترجمه بسیار موقتیاند و در یک تعادل پویا وجود دارند.
اگر توالی آمینواسیدی پروتئین از قبل شناخته شده باشد، میتوان با هضم پروتئین مورد نظر (نمونه مورد آزمایش) با آنزیم تریپسین و آنالیز پپتیدهای حاصله با طیفسنج جرمی و مقایسه جرمهای محاسباتی با جرم مورد انتظار حاصل از هضم فرضی پروتئین با آنزیم مشابه، نوع تغییری که در پروتئین رخ داده را شناسایی نمود. مثلاً در مورد فسفوریلاسیون، افزایش80 دالتونی در جرم پپتید نشان دهنده اضافه شدن یک گروه فسفات به پپتید است. پپتید فسفریله شده (پیتیدی که اضافه جرمی درحدود Da 80 نشان دهد) سپس بوسیله MS/MS مورد آنالیز قرار میگیرد تا جایگاه دقیق فسفوریلاسیون مشخص شود.
حال اگر پروتئین از قبل شناسایی نشده باشد. نمونه پروتئین بعد از هضم آنزیمی، به دو قسمت مساوی تقسیم میشود. یک قسمت را با آنزیم فسفاتاز تیمار میکنند تا تمام گروههای فسفات را حذف نمایند. سپس طیف جرمی هر دو قسمت را با هم مقایسه میکنند تا تغییرات پس از ترجمه نمایان شود(28).
4-مثالهایی از کاربرد های بالینی طیف سنجی جرمی متوالی
1-4 ارزیابی سطچ ویتامین D
ویتامین D در کبد در موقعیت 25 هیدروکسیله میشود و شکل 25OH vitD را ایجاد میکند که فراوانترین فرم ویتامین D در گردش خون است. همچنین این فرم بیشترین طول عمر را دارا میباشد (تقریباً 2ـ1 هفته). غلظت فرم 25OH vitD به عنوان بهترین شاخص سنتز پوستی و دریافت از راه رژیم غذایی محسوب میشود. کاهش مزمن سطح 25OH vitD در کمبود تغذیهای یا استئومالسی و افزایش آن در مسمومیت با ویتامین D شاخص خوبی به حساب میآید. همچنین غلظت 25OH vitDتغییرات فصلی متعاقب مقدار نوری که فرد دریافت میکند را نشان میدهد. بیشترین غلظت آن در اواخر تابستان و کمترین غلظت آن نیز در بهار مشاهده میگردد. روشهای آزمایشگاهی زیادی برای اندازهگیری غلظت ویتامین شامل HPLC/E، رادیوایمونواسی، کمیلومینسس و در نهایت Ms ـ Ms ـLc، وجود دارد(29, 30).
گزارش شده است که مشکلاتی در انجام همه این روشها وجود دارد و مهارت بسیار بالایی برای هرکدام نیاز است. یکی از چالشهایی که وجود دارد تغییرات مقادیر اندازهگیری شده بین روش های موجود میباشد. ولی در بین این روشها، MsـMsـLc کمترین تغییرپذیری را در بین آزمایش با نمونه مشابه دارد. بنابراین MsـMsـLc یک انتخاب مناسب و قدرتمند برای آزمایشگاههای رفرانس محسوب میشود(30, 31).
2-4 کاربرد طیف سنجی جرمی پشت سر هم در برنامه غربالگری نوزادان
هدف اصلی از برنامه غربالگری تشخیص اختلالات بالینی در جهت کاهش پراکندگی و مرگ و میر حاصل از بیماری میباشد. غربالگری نوزادان با روش طیف سنجی جرمی از سال 1960 توسط Guthrie و Susi برای تخمین فنیل آلانین در نمونههای کاغذ صافی جمعآوری شده برای تشخیص بیماری فنیل کتونوری (PKU) ابداع شد(32).
همانطور که توضیح داده خواهد شد برای غربالگری جرمی روشهای جمعآوری نمونه و آنالیز بسیار ساده، سریع و قابل اعتماد خواهد بود. این روش آنقدر موفق بود که در عرض مدت زمان کوتاهی برای غربالگری PKU در تمامی آمریکا و بسیاری دیگر از کشورهای پیشرفته اجرایی شد(33). تا اوایل سال 1990 بیماریهای محدود دیگری نیز به برنامه غربالگری نوزادان اضافه شد. در همان سال 1990 با معرفی تکنیک Tandem mass (Ms/Ms) به آزمایشگاههای غربالگری بیماری الگوی آنالیز یک آنالیت به ازای هر اختلال تغییر کرد. روش Ms /Ms با یک آنالیز مجزا و مدت زمان 2 تا 3 دقیقه لکههای کوچک خون امکان تشخیص آنالیتهای متعدد که اختلالات متعدد متابولیکی (بیشتر از 40 اختلال) را بررسی میکند، فرآهم کرد(34, 35).
در حال حاضر اکثر ایالتهای آمریکا و بسیاری از کشورهای دیگر تکنیک Ms/Ms را به اجرا گذاشتهاند و یا در حال راه اندازی هستند. نمونه خون در نوزادان از طریق پاشنه پا بر روی کاغذهای صافی مخصوص که معمولاً کاغذهای غربالگری نوزاد یا کاغذ گاتری نامیده میشود، جمع آوری میگردد. برای افزایش جریان خون خروجی میتوان ابتدا پاشنه پا را تا دمای °C42 برای چند دقیقه گرم کرد و به دنبال آن تمیز کردن و سوراخ کردن پوست توسط یک لانست استریل به عمق حداکثر mm 2/5 انجام میشود. برای اینکه وضعیت رژیم غذایی نوزادان به طور کامل مورد بررسی قرار گیرد، جمع آوری نمونه باید 24 ساعت بعد از تغذیه دهانی و قبل از مرخص شدن نوزاد از بیمارستان صورت گیرد.
به طور کلی در نوزادان کمتر از 5 روز انجام میشود و اگر نوزادی قبل از 24 ساعت از بیمارستان مرخص شود، برای اینکه نمونه خون نوزاد از دست نرود بهتر است جمعآوری شود و هنگامی که نوزاد برای چک آپ هفته اول به متخصص نوزادان مراجعه میکند، نمونهگیری دوباره تکرار شود. معمولترین طیف جرمی پشت سرهمی که در غربالگری نوزادان استفاده میشود طیف سنج Tripple Quadrapole است(36).
دوره غربالگری بیماریها با اختراع کاغذهای گاتری برای روش مهار باکتریایی[3] (BIA) در سال 1959برای تشخیص سطح فنیلآلانین در افراد مبتلا به فنیل کتونوری (PKU) شروع گردید. در این روش از یک قطره خون خشک شده روی کاغذ صافی مخصوص استفاده میشد. این روش ساده قابل تکرار و ارزان قیمت بود(37). در حال حاضر شناسایی اختلالات توسط برنامه غربالگری نوزادان باروش Ms /Ms به گروههای زیر تقسیم بندی میشود:
1-2-4 شناسایی اختلالات کربوهیدراتها
2-2-4 شناسایی اختلالات آمینواسیدها
3-2-4 اختلالات سیکل اوره
4-2-4 اختلالات اسید ارگانیکها
5-2-4 شناسایی اختلالات اکسیداسیون اسیدهای چرب
6-2-4 شناسایی اختلالات ذخیره لیزوزومی
1-2-4 اختلالات کربوهیدراتها
سه آنزیم در متابویسم گالاکتوز دخیل است که شامل گالاکتوز 1ـ فسفات یوریدیل ترانسفراز (GALT)، UDP گالاکتوز4-اپیمراز(GALE) و گالاکتوزکیناز(GALK) میباشد. گالاکتوزمی کلاسیک به علت کمبود آنزیم GALTاست که منجر به تجمع گالاکتوز1ـ فسفات میشود. مسیر جانبی تجزیه منجر به تولید گالاکتونات و گالاکتیتول میشود که حالت دوم منجر به ایجاد کاتاراکت در چشم میگردد.
کاتاراکت کلاسیک درمان نشده در دوران نوزادی با علائم استفراغ، عدم رشد، زردی، هپاتومگالی، کاتاراکت و سپتیسمی گرم منفی خود را نشان میدهد. در کمبود GALK زودرسترین علامت بیماری کاتاراکت بوده که معمولاً دو طرفه و شروع آن ممکن است از دوران نوزادی باشد و کمبود GALK نادرترین نوع کمبود در متابولیسم گالاکتوز است(38).
غربالگری برای تمام حالات گالاکتوزمی با اندازهگیری فعالیت GALTاز لکههای خون خشک شده و اندازهگیری کمی گالاکتوز1 ـ فسفات سلولهای RBCانجام میشود. نوزادان مبتلا به گالاکتوزمی کلاسیک فعالیت آنزیمی کمتر از 50٪ داشته و همچنین گالاکتوز 1 ـ فسفات در آنها بیشتر از mg /dL20 میباشد که معمولاً در افراد هموزیگوت Q188Rو یا دیگر موتاسیونهای شدید یافت میشود(39).
2-2-4 اختلالات اسیدهای آمینه
این اختلالات به دلیل نقص آنزیمی در مسیر کاتابولیک اسید آمینه ایجاد میشود که منجر به افزایش اختصاصی اسیدآمینه میگردد. این افزایش سطح اسیدآمینه و در بعضی موارد افزایش متابولیتهای مسیرهای جایگزین میتواند منجر به اختلال و چهرههای بالینی متعدد گردد. درمان شامل محدودیت دریافت اسید آمینهها و جیره غذایی است(40).
1-2-2-4 فنیل کتونوری و هیپرفنیلآلانمی
PKU یکی از شناخته شدهترین اختلالات اسید آمینه است و به عنوان اولین علت بیوشیمیایی عقب ماندگی ذهنی شناخته شده است. این بیماری در اثر کمبود آنزیم کبدی فنیلآلانین هیدروکسیلاز در متابولیسم فنیلآلانین ایجاد میشود. این اختلال باعث تجمع فنیلآلانین و سایر متابولیتهای سمی میگردد(41). اگر بیماری درمان نشود علائم بیماری شامل اختلالات نورولوژیک و در نهایت عقب ماندگی ذهنی خواهد بود و اگر در اوایل بیماری و در حالت قبل از بروز علائم تشخیص و درمان شود از عقب ماندگی ذهنی جلوگیری به عمل میآید. اگر چه آنالیزهای اسیدآمینه میتواند توسط Ms /Ms، HPLC و یا BIAانجام شود، ولی در حال حاضر آنالیز Ms/Ms انتخاب اصلی برنامه های غربالگری میباشد.
بیماران با نقص فنیلآلانین هیدروکسیلاز بر اساس سطح فنیلآلانین خود تقسیمبندی میشوند. اگر چه این تقسیم بندی بین پزشکان در نظر گرفته نمیشود ولی به طور کلی بیماران با سطح فنیل آلانین بیشتر از µmol/L 600 افراد مبتلا به PKU در نظر گرفته میشوند و مقادیر 200 به فرم کلاسیک (بیشتر از µmol/L 1200) و ملایم ( 1200 ـ 600 µmol/L) تقسیمبندی میشود. نوزادان و کودکان با این سطح خونی از فنیلآلانین باید محدودیت در جیره غذایی را رعایت کنند. بیماران با سطح فنیل آلانین بین 600 ـ 360 جز دسته هیپرفنیلآلانینمی قرار میگیرند. معمولاً کودکان مبتلا به هیپرفنیلآلانینمی ریسک افزایش سطح PKU به مقادیر ملایم PKU را دارند. پس توصیه شده است که این کودکان تا 1 سالگی تحت کنترل قرارگیرند. حساسیت غربالگری به وسیله Ms/Ms برای PKU حدود 99ـ 98 درصد ذکر شده است(42).
2-2-2-4 اختلال در متابولیسم بیوپترین
بعضی از بیماران با مقادیر افزایش یافته فنیلآلانین دارای نقص در یکی از 4 آنزیم درگیر در بیوسنتز یا تولید تتراهیدروبیوپترین که کوفاکتور بسیاری از آنزیمها از جمله فنیلآلانین هیدروکسیلاز است، میباشد. بیماران مبتلا به اختلال بیوپترین دارای کمبود نوروترانسمیتر بوده و علائمی مثل ناتوانی فکری، تشنج، میکروسفالی و علائم درگیری عصبی را دارند. اگرچه بیماران درمان نشده معمولاً در عرض چند سال میمیرند ولی درمان با مکملهای تتراهیدروبیوپترین ممکن است اثر بخش باشد. مطالعات جنبی برای این اختلال نیازمند آنالیز پتریدین در CSF و ادرار و ارزیابی نوروترانسیترهای CSF میباشد(43).
3-2-2-4 تیروزینمی تیپ I (Hepatorenal)
تیروزینمی تیپ Iبه دلیل نقص در آنزیم فوماریل استواستات هیدرولاز ایجاد میشود. بیماران مبتلا ممکن است در بچگی نارسایی حاد کبدی، نقص در سیستم انعقادی و یا اختلال در توبولهای کلیه و عدم رشد را داشته باشند. بیماران ممکن است بوی کلم پخته شده بدهند. سوکسینیل استون اصلیترین متابولیت سمی در این وضعیت است. یک روش با حساسیت بالا برای غربالگری تیروزینمی تیپ Iاستفاده از تکنیک Ms /Ms برای سوکسینیل استون است. افزایش سریع سوکسینیل استون بعد از تولد نشان دهنده تیروزنیمی تیپ I است که مقادیر بالای µmol/L 87 (نرمال آن کمتر از µmol/L 5) در 12 ساعت اول بعد از تولد نشان دهنده بیماری است(44, 45).
4-2-2-4 تیروزینمی تیپ II
تیروزینمی تیپ IIبه دلیل نقص در آنزیم تیروزین آمینوترانسفراز است. بیماران مبتلا زخمهای قرنیه با التهاب صلبیه، درد در اولین سال زندگی و کراتوزیز پلانتوپالمار بعد از اولین سال زندگی را نشان میدهند. درمان شامل جیره غذایی با نسبت پائین Tyr/Phe است. اگرچه تشخیص تیروزنیمی تیپ IIباروش Ms/ Msانجام میشود ولی گزارشات کمی منتشر شده است. در این بیماری چون مسیر متابولیسمی در اولین مرحله از مسیر مسدود میشود، مقادیر تیروزین سریعاً افزایش مییابد، بدون اینکه افزایش چشمگیری در فنیلآلانین یا متیونین داشته باشیم(46).
5-2-2-4 هموسیستینوری (Homocystinuria )
هموسیستینوری به دلیل نقص در آنزیم سیستاتیونین B-سنتاز میباشد. بیماران درمان نشده از ترومبوامبولی و ظاهر شبیه سندرم مارفان رنج میبرند. درمان شامل بتائین که متیلدار شدن دوباره هموسیستئین به متیونین را تسهیل میکند، میباشد. غربالگری بیماران بر اساس مقادیر بالای متیونین و مقادیر بالای هموسیستئین در تستهای تکمیلی است(42).
6-2-2-4 متیونین آدنوزیل ترانسفراز و اختلالات مرتبط با آن
متیونین آدنوزیل ترانسفراز تیپ I و III ایزوآنزیمهایی هستند که مرحله اول تبدیل متیونین به هموسیستئین را کاتالیز میکنند. بیماران معمولاً مقادیر بالای متیونین را داشته ولی مقادیر هموسیستئین در حد نرمال و یا افزایش کمتری نسبت به بیماران مبتلا به هموسیستینوری دارند و مقادیر sـ آدنوزیل متیونین کاهش یافته است(47).
7-2-2-4 بیماری ادرار شربت افرا (Maple Syrup Urine Disease)
MSUD اختلالی است که با نقص در کمپلکس دهیدروژناز کتواسیدهای شاخهدار که منجر به تجمع لوسین، ایزولوسین و والین به همراه کتواسیدهای خود و همچنین با یک افزایش قابل توجه در آلوایزولوسین همراه است. افزایش مقادیر لوسین منجر به آسیب مغزی و علائم عصبی میشود.
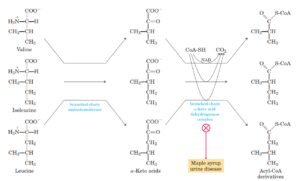 مسیر متابولیسم اسید آمینههای شاخهدار
مسیر متابولیسم اسید آمینههای شاخهدار
غربالگری نوزادان برای MSUD منجر به تشخیص سریع بیماری با مقادیر کم افزایش یافته لوسین پلاسما میگردد و علائم بالینی نیز کمتر است. استفاده از آلوایزولوسین به عنوان آزمایش دوم یک راه حل امیدوار کننده برای کاهش cut off در جهت افزایش قدرت تشخیص واریانتهای MSUD و همچنین در جهت کاهش موارد مثبت کاذب به کار میرود(49).
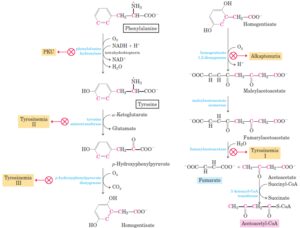
اختلالات مسیر متابولیسمی اسید های آمینه
3-2-4 اختلالات سیکل اوره
سیکل اوره مسئول دفع نیتروژن اضافی ناشی از تجزیه پروتئینها و دیگر مولکولهای حاوی نیتروژن میباشد. 6 آنزیم و 2 ترانسپورتر در این چرخه شناخته شده است. تظاهرات بالینی این اختلالات شامل هیپرآمونمی، استفراغ، بیحالی، کما و مرگ میباشد(42).
1-3-2-4 نقص در اورنیتین ترانس کاربامیلاز ( Ornithine Transcarbamylase)
کمبود OT که یک اختلال وابسته به x می باشد، شایعترین علت اختلالات سیکل اوره بوده و مسئول بسیاری از علائم بیماری است. بیماران مبتلا با کمبود اورنتین ترانسکربامیلاز با سطح پایین مقادیر سیترولین مشخص میشوند. اگر چه غربالگری نوزادان برای کمبود اورنیتین ترانسکربامیلاز (مخصوصاً در نوزادان دختر) قابل اعتماد نیست و ممکن است مقادیر پائین سیترولین با کاهش دریافت پروتئین همراه باشد و یا در موارد بیماریهای رودهای نیز مقادیر سیترولین کاهش یافته است(50).
2-3-2-4 سیترولینمی (citrullinemia)
سیترولینمی به دلیل نقص در آنزیم آرژینوسوکسینات سنتاز بوده و با مقادیر بالای سیترولین مشخص میشود(51).
3-3-2-4 نقص در آرژینوسوکسینات لیاز(Arginosuccinate lyase Deficiency)
نقص در آنزیم آرژینوسوکسینات لیاز نیز با افزایش مقادیر سیترولین مشخص میشود. اگر چه حساسیت این روش تا کنون گزارش نشده است(42).
4-2-4 اختلالات ارگانیک اسیدها
ارگانیک اسیدمیها گروهی از اختلالات هستند که دارای ویژگی مشترک در دفع ترکیبات اسیدی بیوشیمیایی فاقد نیتروژن از طریق ادرار میباشند. بسیاری از این اختلالات در نتیجه نقص عملکرد یک آنزیم درگیر در تجزیه اسید آمینهها بعد از اینکه گروه آمین آنها برداشته شده است، ایجاد میشود. غربالگری نوزادان برای ارگانیک اسیدمیها توسط Ms/Ms برای اندازهگیری سطح و الگوی کارنیتین استرها در لکههای خون خشک شده انجام میشود(42).
1-4-2-4 پروپیونیک اسیدمی
پروپیونیک اسیدمی به دلیل نقص در پروپیونیل ـ coA کربوکسیلاز که آنزیم درگیر در متابولیسم لوسین و ایزولوسین و متابولیتهای دیگر میباشد، ایجاد میگردد. تاکنون گزارشات کمی درباره پروپیونیک اسیدمی در برنامه غربالگری نوزادان منتشر شده است(52). سایر اختلالات مرتبط با ارگانیک اسیدها عبارتند از:
- ایزووالریک اسیدمی
- متیل مالونیک اسیدمی تیپ Mut /Mut
- 3ـ متیل کروتونیل ـ coA ـ کربوکسیلاز
- coA ـ HMG لیاز
- هولوکربوکسیلاز سنتاز
- گلوتاریک اسیدوری تیپ I
- ایزوبوتیریل گلیسینوری
- مالونیک اسیدوری
5-2-4 اختلالات اکسیداسیون اسیدهای چرب
تمام اختلالات اکسیداسیون اسیدهای چرب با اختلال در تولید انرژی همراه است. به طور کلی علائم و نشانهها شامل عدم تحمل گرسنگی، هیپوگلیسمی هیپوکتونیک و یا غیرکتوتیک، بیحالی، میوپاتی، نقص عملکرد کبدی، گاهی نیز به شکل سندرم شبیه سندرم ری خود را نشان میدهد.
1-5-2-4 کمبود (short chain acyl_coA dehydrogenase) SCAD
مارکرهای تشخیص این کمبود و افزایش مقادیر اسیدهای چرب C4 میباشد(40).
2-5-2-4 کمبود (Medium chain acyl-coA dehydrogenase) MCAD
تا پیش از ابداع غربالگری نوزادان کمبود MCAD علت 19 تا 25 درصد مرگ و میرها و همچنین به عنوان یک ریسک فاکتور مهم بیماریی مثل تأخیر در رشد، مشکلات رفتاری، ضعف عضلانی و عدم رشد بوده است .از زمان ایجاد برنامه غربالگری نوزادان تعداد بیماران مبتلا به کمبود MCAD دو برابر شده است(53). بسیاری به این بیماری، سندرم شبهری یا سندرم مرگ ناگهانی شیر خواران میگفتند. مارکرهای تشخیص این بیماری افزایش مقادیر اسیدهای چرب C6 و C8 و C10 میباشد(40).
3-5-2-4 کمبود (Very Long Chain Acyl–coA Dehydrogenase) VLCAD
مارکرهای تشخیص این کمبود افزایش مقادیر اسیدهای چرب C14:2 ,C14:1 ,C14 میباشد(40).
4-5-2-4 کمبود (Carnitine Polmitoyl Transferase Typ I) CPT-1
کمبود CPT -1 تنها اختلال اکسیداسیون اسیدهای چرب است که با افزایش مقادیر کارنیتین بروز مییابد(54).
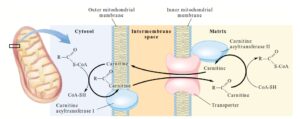
عبور اسید چرب توسط چرخه کارنیتین
6-2-4 اختلالات ذخیرهای لیزوزمی
اختلالات ذخیرهای لیزوزمی گروه هتروژنی از تقریباً 50 بیماری است که در آنها نقصی در عملکرد لیزوزمی شامل نقصهای آنزیمی، رسپتورهای آنزیم، فعال کنندههای پروتئینی و یا ترانسپورترها میباشد. معمولاً این اختلالات باعث تجمع سوبستراهایی میشود که منجر به تخریب سلولی و نقص در عملکرد بافتی میشود. تمام اختلالات لیزوزومی به جز سندرم Hanter، بیماری Fabry و بیماری Danon که وابسته به x هستند، اتوزومی مغلوب میباشند(55).
غربالگری نوزادان برای اختلالات ذخیرهای لیزوزومی در اوایل راه میباشد و در حال حاضر هیچ کدام از اختلالات جزو برنامه غربالگری ژنتیک پزشکی کالج آمریکایی نیست. روشهایی که برای تشخیص این اختلالات به کاربرده میشود شامل Ms/Ms و روشهای فلوریمتری میباشد(56).
3-4 نقش Tandem MS در تشخیص بیومارکرهای سرطانی
در طی سالهاي اخیر توجه زیادي به نقش بیومارکر در تشخیص سرطان در مقطع مطالعات کلینیکی شده است(57). بیش از دو سوم مبتلایان به ﺳﺮﻃﺎن ﺗﺨﻤﺪان در مراحل پیشرفته سرطانشان آگاهی پیدا میکنند. این سرطان تا زمانی که به مرحله بدخیمی نرسیده است، علائم مشخصی از خود نشان نمیدهد و وقتی به مرحله پیشرفته خود رسید، امکان درمان آن مشکلتر میشود. اما اگر در همان مراحل اولیه کنترل شود، شانس بیمار براي بقاء به بیش (stage1) از 5 سال خواهد رسید(58). طیفسنجی روشی مناسب براي آنالیز دادههاي طیفسنجی جرمی بیولوژیکی خصوصاً سرطان است.
دقت این تکنیک براي شناسایی بیومارکرها بسیار بالا است. در مطالعهای در سال 2012 مجموعهای از تومور مارکرهای سرطان ریه به روش LC-MS/MS بررسی گردید. این روش قادر به شناسایی تغییرات حتی در پروتئینهاي بسیار کوچک میباشد(59). همچنین در مطالعهای که در سال 2009 توسط Soleiman و همکاران صورت گرفت با استفاده از تکنیکcapillary electrophoresis-tandem mass spectrometry بر روی نمونههای ادراری تومور مارکرهای مربوط به سرطان پروستات تشخیص داده شد(60).
منابع:
- Janeza T. Tandem Mass Spectrometry – Applications and Principles. croatia: InTech; 2012.
- سلیمانی. پ, سیدمتین. ن, تقوی مقدم، ع.ا. طیف سنجی جرمی. تهران: جهاد دانشگاهی 1372.
- Thomson JJ. Rays of positive electricity and their application to chemical analyses. London: Green and Co; 1921.
- Gross JH. Mass Spectrometry: A Textbook. Berlin: Springer-Verlag; 2004.
- Wilson K, Walker J. Principles and Techniques of Biochemistry and Molecular Biology. seven ed ed. UK: Cambridge University Press; 2010.
- Pavia DL, Gary ML, George SK, James RV. Introduction to Spectroscopy. fourth edition ed. Washington, United States of America: Department of Chemistry Western Washington University Bellingham; 2008.
- Gross JH. Mass Spectrometry. Second Edition ed. Berlin Springer-Verlag 2010.
- Tania P, Elena P, Francisco JL, Félix H, Wilfried MAN. Use of soft and hard ionization techniques for elucidation of unknown compounds by gas chromatography/time-of-flight mass spectrometry. Rapid CommunMass Spectrom 2011;25:1589-99.
- Edmond dH, Vincent S. Mass Spectrometry Principles and Applications. Third Edition ed. 2007, editor. USA: John Wiley & Sons Ltd.
- Afsharypour S. Instrumental Methode of Analaysis. Isfahan: Isfahan university of medical science; 1372.
- Munson MSB. Development of Chemical Ionization Mass Spectrometry. Int J Mass Spectrom 2000;200:243-51.
- Olson KL, Rinehart KL. Field Desorption, Field Ionization, and Chemical Ionization Mass Spectrometry. Methods Carbohyd Chem. 1993;9(143-164).
- Lattimer RP, Schulten HR. Field Ionization and Field Desorption Mass Spectrometry Past, Present, and Future. Anal Chem 1989;61(1201A-1215A).
- Heumann KG, Schindlmeier W, Zeininger H, Schmidt M. Application of an Economical and Small Thermal Ionization Mass Spectrometer for Accurate Anion Trace Analyses. Zeitschrift für Analytische Chemie 1985;320:457-62.
- Devienne FM, Roustan JC. Fast Atom Bombardment” – A Rediscovered Method for Mass Spectrometry. Org Mass Spectrom 1982;17:173-81.
- Lane CS. Mass spectrometry-based proteomics in the life sciences. Cell Mol Life Sci 2005;62:848-69.
- Lin D, Tabb DL, Yates JR. Large-scale protein identification using mass spectrometry. Biochimica et Biophysica Acta 2003;1646:1-10.
- Ashcroft AE. An Introduction to Mass Spectrometry. Mass Spectrometry Facility Manager. Building, the University of Leeds.: Astbury Centre for Structural Molecular Biology; 2002 .
- Griffith WJ, Andreas PJ, Suya LIU, Rai KD, Yuqin W. Electrospray and tandem mass spectrometry in biochemistry. Biochem J 2001;355(545-561).
- Erin JF, Lee KH. An Introduction to Mass Spectrometry Applications in Biological Research. Biochemistry and Molecular Biology 2004;32(2):93-100.
- Gharechahi J, Naghavi MR, Alizadeh H. Mass Spectrometry and Its Application in Proteomics. Modern Genetics 2009;3(4):5-28.
- Roboz J. Mass Spectrometry in cancer research. The Mount Sinai School of Medicine New York CRC PRESS; 2002.
- Yates JR. Mass spectrometry and the age of the proteome. J Mass Spectrom. 1998;33:1-19.
- Wilkins MR, Sanchez JC, Gooley AA, Appel RD, Humphery-Smith I, Hochstrasser DF, et al. Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotechnol Genet Eng Rev 1995;13:19-50.
- Junmin P, Gygi SP. Proteomics: the move to mixtures. J Mass Spectrom 2001;36:1083-91.
- Reid GE, McLuckey SA. ‘Top-down’ protein characterization via tandem mass Spectrometry. J Mass Spectrom 2002;37:663-75.
- Sara TH, Hodge K, Lamond AI. Dynamic Proteomics: Methodologies and Analysis Functional Genomics: InTech; 2012. p. 181-200.
- Jawon S, Kong-Joo L. Posttranslational Modifications and Their Biological Functions: Proteomic Analysis and Systematic Approaches. Journal of Biochemistry and Molecular Biology 2004;3(1):35-44.
- Higashi T, Awada D, Shimada K. Simultaneous determination of 25-hydroxyvitamin D-2 and 25-hydroxyvitamin D-3 in human plasma by liquid chromatographytandem mass spectrometry employing derivatization with a Cookson-type reagent. Biol Pharm Bull 2001;24:738-43.
- Binkley N, Krueger D, Cowgill CS, Plum L, Lake E, Hansen KE, et al. Assay variation confounds the diagnosis of hypovitaminosis D: a call for standardization. J Clin Endocrinol Metab 2004;89:3152-7.
- Singh RJ. Are clinical laboratories prepared for accurate testing of 25-hydroxy vitamin D? Clin Chem 2008;54:221-3.
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:338-43.
- Paul DB. The history of phenylketonuria screening in the US. In: Watson MS, editor. Promoting safe and effective genetic testing in the United States: final report of the task force on genetic testing. Bethesda. National Institutes of Health 1997.
- Rhead WJ, Irons M. The call from the newborn screening laboratory: frustration in the afternoon. Pediatr Clin North Am 2004;51(3):803-18.
- Chace DH. Mass spectrometry in the clinical laboratory. Chem Rev 2001;101(2):445-77.
- Uttam G, Majed D. Expanded newborn screening of inherited metabolic disorders by tandem mass spectrometry: Clinical and laboratory aspects. Clinical Biochemistry. 2006;39:315-32.
- Levy HL. Lessons from the past—looking to the future. Newborn screening. Pediatr Ann. 2003;32:505-8.
- Epimerase deficiency galactosemia. In: GeneReviews at GeneTests Medical Genetics Information Resource. . 1997-2012. [database on the Internet]. 2011 [cited http://www.genetests.org.].
- Galactosemia. In: GeneReviews at GeneTests Medical Genetics Information Resource [database on the Internet]. 2011 [cited http://www.genetests.org.].
- Inderneel S, Deborah M. Newborn Screening. Critical Reviews in Clinical Laboratory Science 2009;46(2):55-82.
- Jervis GA. Phenylpyruvic oligophrenia deficiency of phenylalanine-oxidizing system. Proc Soc Exp Biol Med 1953;82:514–5.
- Angela S, Christina L, Derek AW. Expanded Newborn Screening for Inborn Errors of Metabolism Overview and Outcomes. Advances in Pediatrics 2012;59:209-45.
- Ponzone A, Spada M, Ferraris S, et al. Dihydropteridine reductase deficiency in man: from biology to treatment. Med Res Rev 2004;24(2):127-50.
- Hutchesson AC, Hall SK, Preece MA, et al. Screening for tyrosinaemia type I. Arch Dis Child Fetal Neonatal Ed 1996;74(3):191-4.
- Schlump JU, Mayatepek E, Spiekerkoetter U. Significant increase of succinylacetone within the first 12 h of life in hereditary tyrosinemia type 1. Eur J Pediatr 2010;169(5):569-72.
- Scott CR. The genetic tyrosinemias. Am J Med Genet 2006;142(2):121-6.
- Chamberlin ME, Ubagai T, Mudd SH, et al. Methionine adenosyltransferase I/III deficiency: novel mutations and clinical variations. Am J Hum Genet 2000;66(2):347-55.
- Lehninger A, David L, Michael M. Lehninger Principles of Biochemistry. Fourth Edition ed. 2008.
- Simon E, Flaschker N, Schadewaldt P, et al. Variant maple syrup urine disease (MSUD)—the entire spectrum. J Inherit Metab Dis 2006;29(6):716-24.
- Cavicchi C, Malvagia S, Marca G, et al. Hypocitrullinemia in expanded newborn screening by LC-MS/MS is not a reliable marker for ornithine transcarbamylase deficiency. J Pharm Biomed Anal 2009;49(5):1292-5.
- Sander J, Janzen N, Sander S, al. e. Neonatal screening for citrullinaemia. Eur J Pediatr 2003;162(3):417-20.
- Yorifuji T, Kawai M, Muroi J, et al. Unexpectedly high prevalence of the mild form of propionic acidemia in Japan: presence of a common mutation and possible clinical implications. Hum Genet 2002;111(2):161-5.
- Iafolla AK, Thompson RJ, Roe CR. Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr 1994;124(3):409-15.
- Bonnefont JP, Djouadi F, Prip-Buus C, al. e. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 2004;25:495-520.
- Wang RY, Bodamer OA, Watson MS, al. e. Lysosomal storage diseases: diagnostic confirmation andmanagement of presymptomatic individuals. Genet Med 2011;13(5):457-84.
- Reuser AJ, Verheijen FW, Bali D, al. e. The use of dried blood spot samples in the diagnosis of lysosomal storage disorders – Current status and perspectives. Mol Genet Metab 2011;104(1-2).
- Honda K, Ono M, Shitashige M, Masuda M, R N, Kamita M, et al. Proteomic approaches to the discovery of cancer biomarkers for early detection and personalized medicine. . Japan J Clin Oncol 2013;43:103-9.
- Wulfkuhle JD, Liotta LA, Petricoin EF. Proteomic applications for the early detection of cancer. Nat Rev Cancer 2003;3(267-275).
- Xuemei Z, Brian L H, Ting Z, Thomas P C, Mai Sun MS, al. e. Lung Cancer Serum Biomarker Discovery Using Label Free LC-MS/MS. J Thorac Oncol 2012;6(4):725-34.
- Soliman LC, Hui Y, Hewavitharana AK, Chen DD. Monitoring potential prostate cancer biomarkers in urine by capillary electrophoresis-tandem mass spectrometry. J Chromatogr 2009;7(1267):162-9.
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام