
سندرم پیری زودرس هاچینسون گیلفورد
Hutchinson–Gilford progeria syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، دکتر روشنک سامبرانی (متخصص ژنتیک مولکولی)، سپیده باستانی (کارشناس ارشد بیوشیمی)،
دلشاد عبداللهنیا (کارشناس ارشد بیوتکنولوژی)، الهام احمدی (دانشجوی کارشناسی ارشد بیوتکنولوژی پزشکی)

نگارنده مسئول: شاهین اسعدی (Molecular Geneticist)
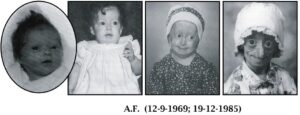
سندرم پیری زودرس هاچینسون گیلفورد (HGPS) یک بیماری نادر و کشنده ژنتیکی است که از دوران کودکی با ویژگیهای قابلتوجه شبیه پیری زودرس همراه است. کودکان مبتلا به سندرم پیری زودرس، معمولاً ظاهر طبیعی در دوران شیرخواری دارند. در حدود 9 تا 24 ماهگی، کودکان مبتلا به این بیماری تحتتأثیر اولین علائم بیماری قرار میگیرند که شامل کوتاهی قد و کاهش وزن است و بهتدریج تأخیر عمیق در رشد و نمو را تجربه خواهند کرد، همچنین این کودکان دارای فرم صورت متمایز، چهره نامتناسب کوچک در مقایسه با توسعه سر، فک تکاملیافته ناقص (micrognathia) و ناهنجاری شکل دندانها هستند. این اختلال ژنتیکی معمولاً 1 در 18 میلیون تولد اتفاق میافتد. متوسط عمر مبتلایان به سندرم پیری زودرس حدود 20 سال است و معمولاً این مبتلایان در اوایل بیست سالگی میمیرند. این سندرم اولین بار در سال 1886 توسط دکتر جاناتان هاچنسون گزارش گردید، اما بهصورت کاملتر در سال 1897 توسط دکتر هاستینگز گیلفورد تشریح و بهصورت اشتراکی بنام سندرم هاچینسون گیلفورد نامگذاری گردید.

علائم و نشانههای بالینی سندرم هاچینسون گیلفورد
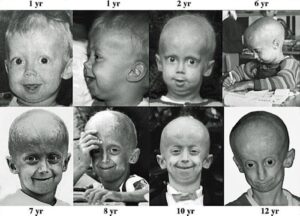
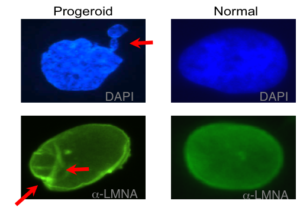
اولین علائم سندرم پیری زودرس معمولاً در پوست بهصورت اسکلرودرمی نمایان میشود. این علائم معمولاً در 18 تا 24 ماهگی کودکان مبتلا به سندرم پیری زودرس آشکار میگردد. رشد محدود، ریزش کامل موهای بدن، چهره کوچک با فک کمعمق و بینی باریک از نشانه سندرم هاچینسون گیلفورد است. این علائم در کودکی بهصورت پیشرونده رخ میدهند و سپس باعث چروکیده شدن پوست، نارسایی کلیهها، از دست دادن بینایی و مشکلات قلبی عروقی میگردد. سختی پوست و سفتی تنه اندامها از نشانههای رایج سندرم هاچینسون گیلفورد است. کودکان مبتلا به سندرم پیری زودرس با 10 سال سن، ارتفاع قامتی مشابه کودکان 3 ساله دارند. شکل جمجمه معمولاً نامتعارف است و همچنین این کودکان دارای گوشهای کوچک و لبهای نازک و بدریختی دندانها و پوسیدگی زودرس دندانها میباشند.
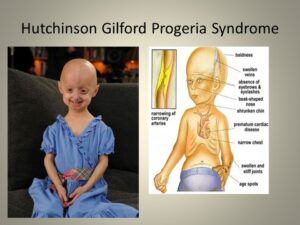
کودکان مبتلا به HGPS دارای وریدهای آشکار در مناطق خاصی از بدن بخصوص در پوست سر و ران هستند. علاوه بر این، لکههای قهوهای رنگ در پوست این کودکان پدیدار میشود که ممکن است با افزایش سن و در معرض قرار گرفتن نور خورشید، این لکههای قهوهای رنگ بیشتر توسعه یابند. کودکان مبتلا به سندرم پیری زودرس، معمولاً دارای ناخنهای ناقص مانند ناخن پا، زرد رنگ، نازک، شکننده، منحنی هستند و یا ممکن است در بعضی انگشتان هیچ ناخنی وجود نداشته باشد. همچنین این کودکان دارای نقص اسکلتی هستند و معمولاً شکل جمجمه سر این کودکان به شکل گنبد است. علاوه بر این، کودکان مبتلا به سندرم هاچینسون گیلفورد استخوانهای دست و پای بلند و نازک و شکننده و قفسه سینه گلابی شکل و استخوانهای شکننده در بازوها را دارند، همچنین علائمی مانند صدای با تن بالا در سنین نوجوانی، فقدان پستان یا نوک پستان، فقدان بلوغ جنسی، اختلال شنوایی و بینایی در مبتلایان سندرم پیری زودرس مشهود است.
از علائم دیگری سندرم پیری زودرس میتوان به اختلالات عروق کرونری و شریانهای لنفاوی اشاره کرد. کودکان مبتلا به این سندرم معمولاً در سنین پنج سالگی، دردهای قفسه سینه به علت کمبود اکسیژنرسانی به عضله قلب را تجربه میکنند.




ژنتیک مولکولی سندرم هاچینسون گیلفورد
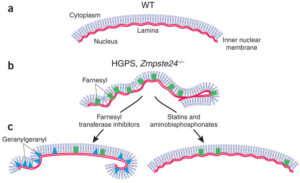
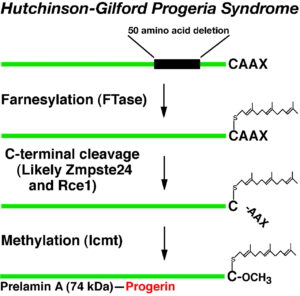
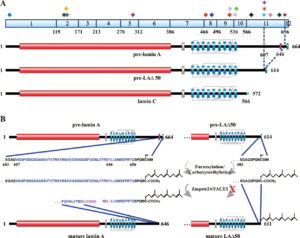
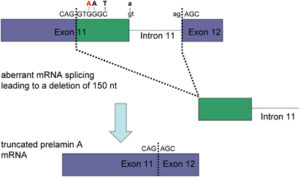
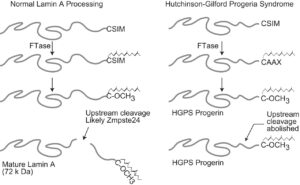
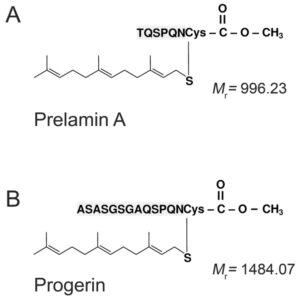
ژنتیکدانان علت سندرم HGPS را یک حرف غلط املایی در ژن LMNA در کروموزوم شماره 1 تعیین کردهاند. ژن LMNA که کدکننده پروتئین Perlamin A میباشد در بازوی بلند کروموزوم شماره 1 بهصورت 1q22 مستقر است و یک جزء کلیدی در ساختار سیتوپلاسم سلول میباشد. یک جهش نقطهای در موقعیت 1824 ژن LMNA که در آن سیتوزین با تیمین جایگزین شده است منجر به سندرم پیری زودرس میشود. این جهش در توالی ‘5 ژن LMNA در اگزون شماره 11 رخ میدهد که منجر به کوتاه شدن توالی RNA پیامبر میشود که در طی آن، پروتئین Prelamin A نمیتواند از آنزیم فارنسیل ترانسفراز جدا شود. آنزیم فارنسیل ترانسفراز، آنزیمی است که بهطور موقت اجازه میدهد تا Prelamin A به لبه هستهای سلول متصل شود. از آنجا که گروه فارنسیل طی این جهش نمیتواند حذف شود، پروتئین Prelamin A بهطور غیرطبیعی بهعنوان پروتئین Progerin سنتز خواهد شد که بهصورت دائمی به لبههای هستهای سلول چسبیده باقی میماند، در نتیجه لامین A تشکیل نخواهد شد و بدون لامین A، لایه هستهای قادر به ارائه پوشش هستهای با پشتیبانی از ساختاری مناسب، نخواهد بود. بدلیل اینکه پشتیبانی لایههای هستهای بهطور معمول، سازماندهی کروماتین در طول تقسیم میتوز را فراهم میکند، تضعیف لایه هستهای، توانایی تقسیم سلول را محدود خواهد کرد.
تا امروز بیش از 1400SNP یا پلیمورفیسم تکنوکلئوتیدی در ژن LMNA شناخته شده است. این پلیمورفیسمها میتوانند در تغییرات mRNA، پیرایش ترجمه کدونها و سنتز سطحی پروتئین با جهشهایی مانند: Arg471Cys، Arg482Gln، Arg527Leu، Arg527Cys و Ala529Val ایفای نقش کنند.
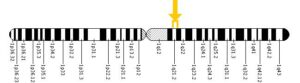
بیماری HGPS بهصورت اتوزومال غالب به ارث میرسد و تنها یک کپی از ژن معیوب میتواند این سندرم را بروز دهد.
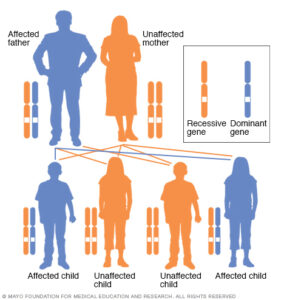
والدینی که یک کودک مبتلا به سندرم پیری زودرس را دارند، شانس بیشتری به میزان 3-2% برای داشتن فرزند دوم نیز به سندرم پیری زودرس خواهند داشت. این افزایش شانس، بخاطر یک بیماری به نام موزائیسم است که در آن، پدر و مادر جهش ژنتیکی برای پیری زودرس در بخش کوچکی از سلولهای خود را دارند، اما با این حال خود پدر و مادر مبتلا به پیری زودرس نیستند.
علت زمینهای خاص از شتاب پیری مرتبط با HGPS هنوز ناشناخته است. بسیاری از محققان معتقدند که روند پیری با توجه به آسیب سلولی و متابولیک فرایندهای درون سلولی بدن غیرطبیعی است. برخی از محققان معتقدند که کاهش فعالیت آنزیمهای خاصی ممکن است نقشی در ایجاد شتاب پیری زودرس در افراد مبتلا به HGPS داشته باشد. در تحقیقی که سلولهای پوست (فیبروبلاست) بیماران مبتلا به HGPS با سلولهای پوست افراد سالم، مقایسه شد، نشان داده شد که سطح فعالیت آنزیمهای آنتیاکسیدان مانند گلوتاتیون پراکسیداز و کاتالاز در فیبروبلاست مبتلایان HGPS بهطور قابلتوجهی پائینتر از سطح همین آنزیمها در فیبروبلاست افراد سالم است.
افراد در معرض سندرم پیری زودرس
HGPS یک اختلال ژنتیکی بسیار نادر است که مردان و زنان را بهطور مساوی و تقریباً تمام نژادها را بهطور مساوی درگیر میکند. در ژانویه 2014 حدود 200 مورد از مبتلایان HGPS در سراسر جهان گزارش شده است. برآوردها نشان میدهد که شیوع HGPS یک در 18 میلیون تولد اتفاق میافتد، بنابراین با استناد به فرکانس شیوع این بیماری میتوان نتیجه گرفت که حدود 400-350 کودک مبتلا به HGPS در سراسر جهان زندگی میکنند.
مسیر درمانی سندرم پیری زودرس
هیچ درمان مؤثری برای درمان HGPS وجود ندارد، اما میتوان با جراحی قلب از عوارض قلبی عروقی کاست و یا با مصرف آسپرین در دوز پائین میتوان اختلالات عروق کرونری را کاهش داد و همچنین برای کودکان مبتلا به HGPS میتوان رژیم غذایی با انرژی بالا تجویز نمود. هورمونتراپی جهت افزایش هورمون رشد و نمو، استفاده از مورفولینوس بهمنظور کاهش تولید Progerin برای جلوگیری از اختلالات قلبی و عروق کرونری و همچنین الیگونوکلئوتیدهای آنتیسنس مورفولینو برای اگزونهای جهشیافته 11 و 12 در محل اتصال به مولکول mRNA، نیز در تخفیف رنج بیماران مبتلا به HGPS بکار گرفته میشود.
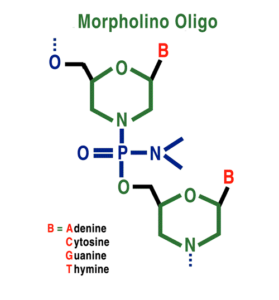
از داروهای ضدسرطانی مانند مهارکنندههای فارنسیل ترانسفراز نیز برای کاهش تولید Progerin و حذف آن از غشای هستهای بهصورت اتوفاژی، استفاده شده است. همچنین لونافارنیب نیز برای این منظور بکار رفته است ولی به دلیل سمی بودن این داروها، سازمان FDA آمریکا آنها را تائید نکرده است و بهمین علت از این داروها در تحقیقات بالینی سندرم پیری زودرس استفاده میگردد.
Reference:
- James، William; Berger، Timothy; Elston، Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 574.
- Rapini، Ronald P.; Bolognia، Jean L.; Jorizzo، Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby.
- Ramírez CL، Cadiñanos J، Varela I، Freije JM، López-Otín C (2007). “Human progeroid syndromes، aging and cancer: new genetic and epigenetic insights into old questions”. Cell. Mol. Life Sci. 64: 155–70.
- Ewell Steve Roach; Van S. Miller (2004). Neurocutaneous Disorders. Cambridge University Press. p. 150.
- Hutchinson J (1886). “Case of congenital absence of hair، with atrophic condition of the skin and its appendages، in a boy whose mother had been almost wholly bald from alopecia areata from the age of six”. Lancet I (3272): 923.
- De Sandre-Giovannoli، A.; Bernard، R.; Cau، P.; Navarro، C.; Amiel، J.; Boccaccio، I.; Lyonnet، S.; Stewart، CL.; et al. (Jun 2003). “Lamin a truncation in Hutchinson-Gilford progeria”. Science 300 (5628): 2055.
- Zirn B، Kress W، Grimm T، Berthold LD; et al. (2008). “Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia، progeria، and rigid spine muscular dystrophy”. Am J Med Genet A 146A (8): 1049–1054.
- Cao H، Hegele RA; Hegele (2002). “Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy”. Hum. Molec. Genet. 9 (1): 109–12.
- Al-Haggar M، Madej-Pilarczyk A، Kozlowski L، Bujnicki JM، Yahia S، Abdel-Hadi D، Shams A، Ahmad N، Hamed S، Puzianowska-Kuznicka M; Madej-Pilarczyk; Kozlowski; Bujnicki; Yahia; Abdel-Hadi; Shams; Ahmad; Hamed; Puzianowska-Kuznicka (2012).
- Garg A، Cogulu O، Ozkinay F، Onay H، Agarwal AK; Cogulu; Ozkinay; Onay; Agarwal (2005). “A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia”. J. Clin. Endocrinol. Metab. 90 (9): 5259–64.
- Corcoy R، Aris A، de Leiva A (1989). “Fertility in a case of progeria”. Am. J. Med. Sci. 297: 383–4.
- Gordon، Leslie B.; Cao، Kan; Collins، Francis S. (2012). “Progeria: Translational insights from cell biology”. J Cell Biol 199 (1): 9–13.
- Lans H، Hoeijmakers JH; Hoeijmakers (2006). “Cell biology: ageing nucleus gets out of shape”. Nature 440 (7080): 32–4.
- Redwood AB، Perkins SM، Vanderwaal RP، Feng Z، Biehl KJ، Gonzalez-Suarez I، Morgado-Palacin L، Shi W، Sage J، Roti-Roti JL، Stewart CL، Zhang J، Gonzalo S (2011).
- Liu B، Wang J، Chan KM، Tjia WM، Deng W، Guan X، Huang JD، Li KM، Chau PY، Chen DJ، Pei D، Pendas AM، Cadiñanos J، López-Otín C، Tse HF، Hutchison C، Chen J، Cao Y، Cheah KS، Tryggvason K، Zhou Z (2005). “Genomic instability in laminopathy-based premature aging”. Nat. Med. 11 (7): 780–5.
- Bernstein H، Payne CM، Bernstein C، Garewal H، Dvorak K (2008). Cancer and aging as consequences of un-repaired DNA damage. In: New Research on DNA Damages (Editors: Honoka Kimura and Aoi Suzuki) Nova Science Publishers، Inc.، New York، Chapter 1، pp. 1-47.
- Singh V (2010). “Reflections: neurology and the humanities. Description of a family with progeria by Charles Dickens”. Neurology 75 (6): 571.
- Robin Marantz Henig (January 30، 2005). “Racing with Sam”. The New York Times. Retrieved July 18، 2016.
- Gordon، LB. The Premature Aging Syndrome Hutchinson-Gilford Progeria: Insights into Normal Aging in: Brocklehurst’s Textbook of Geriatric Medicine and Gerontology، Seventh Edition. 2010:66-72.
- Brown WT. Progeria. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia، PA. 2003:724-5.
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام