
بیماری نوروفیبروماتوز
فضلا… میرزایی نسب1 – دکتر محمد فرزام1
1: (بنیاد بیماریهای نادر و وراثت نسل سالم)
هرچند در ابتدا در متون پزشکی قرن هجدهم به علائم بالینی نوروفیبروماتوز (NF) اشاره شده است، اما از نظر تاریخی این بیماری معمولاً با نام فون رکلین هاوسن همراه است. این آسیبشناس آلمانی در سال 1882 واژه نوروفیبروما را ابداع کرد. نوروفیبروماتوز یکی از رایجترین اختلالات ژنتیکی شناخته شده است. این بیماری زمانی شهرت عمومی یافت که اظهار شد ژوزف مریک، مرد فیلی، احتمالاً به این بیماری مبتلا بوده است. بههرحال (با مطالعه دوباره اسکلت و عکسهای مریک) امروزه بسیاری عقیده دارند که او نوروفیبروماتوز نداشته است و بجای آن به بیماری بسیار کمیابتری به نام نشانگان پروتئوس مبتلا بوده است.
نوروفیبروماتوز (Neurofibromatosis) یک بیماری ژنتیکی است که اعصاب و پوست را گرفتار میکند. در این بیماری تومورهای خوشخیم غیرسرطانی در مسیر اعصاب رشد میکنند و این رشد موجب بروز مشکلاتی در پوست و استخوانها میشود.
دو نوع اصلی نوروفیبروماتوز NF1 و NF2 وجود دارد؛ هر دو بیماری بویژه NF2 را میتوان جزء سرطان وارثتی (فامیلی) طبقهبندی کرد. بروز NF1 رایجتر است و تقریباً برابر با 1 در 3000 تولد است. بروز تقریبی NF2 یک در 35000 بوده و شیوع آن در حدود 1 در 200.000 است.
ویژگیهای بالینی
برجستهترین علائم NF1 زخمهای کوچک پوستی رنگدانهیی بنام لکههای شیرقهوهای (CAL) و تکههای گوشتی نرم کوچک به نام نوروفیبروماتا هستند. لکههای شیرقهوهای ابتدا در اوایل کودکی ظاهر شده و تا زمان بلوغ، هم در اندازه و هم در تعداد افزایش مییابند. دستکم 6 لکه شیرقهوهای به قطر دستکم 5 میلیمتر برای تشخیص در دوران کودکی لازمند. ککمکهای زیربغلی و یا کشالهای نیز باید وجود داشته باشد. نوروفیبروماتا تومورهای خوشخیمی هستند که بهطور معمول در پوست ایجاد میشوند. این تومورها در دوران بلوغ پس از آن ظاهر شده و با افزایش سن تعداد آنها افزایش مییابد.
دیگر یافتههای بالینی در NF1 عبارتند از: ککمکی شدن زیربغل، ماکروسفالی (سربزرگ) نسبی و غدهها یا گرههای لیش. این علائم، هامارتومهای کوچک پیگمانتهی بیضرر در عنبیه هستند. معمولترین مشکل که در یکسوم موارد کودکی رخ میدهد، تأخیر در یادگیری (مسائل غیرکلامی) است که بهصورت خفیفی دیده میشود، البته در بسیاری بیماران این مشکل در دوران مدرسه برطرف میشود. بیشتر افراد NF1 زندگی طبیعی سالمی داشته و بهندرت بواسطهی بیماری خود دچار رنج و عذاب میشوند، با وجود این، شمار اندکی از بیماران یک یا چند شکل اصلی مثل صرع، تومورهای سیستم عصبی مرکزی و اسکولیوز را بروز میدهند (شکل 1).
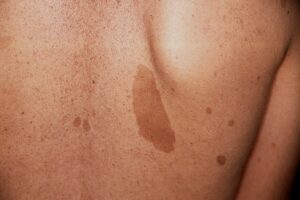
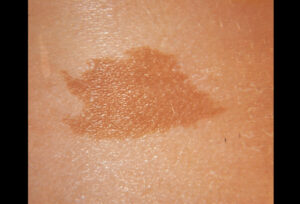
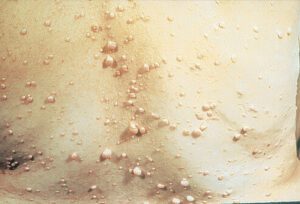
شکل 1: علائم بالینی بیماری نوروفیبروماتوز
ژنتیک بیماری
NF1 توارث غالب اتوزومی نشان داده و تا سن 5 سالگی نفوذپذیری کامل دارد. آثار بیماری بسیار متغیر است و اعضای مبتلای یک خانواده میتوانند تفاوتهای کاملاً مشخصی در شدت بیماری نشان دهند. علائم بیماری در دوقلوهای یکسان مبتلا، معمولاً بسیار شبیه هم هستند، بنابراین بروز متغیر در اعضای خانواده که جهش یکسانی دارند احتمالاً به دلیل تأثیرات ژنهای تعدیلکننده در لوکوسهای دیگر است. در حدود 50 درصد موارد NF1 به دلیل جهشهای جدید رخ میدهد، بهطوریکه این بیماری هیچ سابقهای در خانواده و فامیل بیمار ندارد. در این موارد بیماری به علت جهش در ژن خود فرد بوجود آمده و ربطی به والدین ندارد، ولی وقتی این بیماری حتی بهصورت جهش ژنی در فرد ایجادشود، میتواند به فرزندان او هم منتقل شود.
با توجه به توارث اتوزومال غالب بودن بیماری، اینکه از والدین سالم (غیرمبتلا)، کودکان مبتلا به NF1 متولد شده باشد، گزارشهای اندکی وجود دارد. این حالت احتمالاً بر اثر جهش در سلولهای جنسی والدین است. موزاییسم سوماتیکی در بیماری NF1 میتواند سبب شود تا بیماری تنها در یک بخش از بدن بروز کند، این وضعیت NF Segmental نامیده میشود.
ژن نوروفیبروماتوز تیپ 1 و محصول پروتئین آن:
موقعیت کروموزومی ژن NF1 در سال 1987 بر روی کروموزوم 17 و در نزدیکی سانترومر آن تعیین شد. ژن NF1 پروتئینی به نام نوروفیبرومین را کد میکند. این پروتئیــــــــــــــــن با پروتئین GAP (GTPase-activating protein) (که نقش اصلی در مسیرهای انتقال پیام دارد) هومولوژی ساختاری داشته که با کاهش فعالیت تنظیمی RAS در انتقال سیگنال نقش مهمی ایفا میکند. از دست رفتن هتروزیگوتی نشانگرهای کروموزوم 17 در چندین تومور بدخیم بیماران NF1 و در شمار اندکی از نوروفیبرومهای خوشخیم مشاهده شده است. این مشاهدات نمایانگر آن است که ژن NF1 بهعنوان یک فرونشاننده یا سرکوبگر تومور عمل میکند.
ژنهای دیگری از جمله TP53 واقع بر روی بازوی کوتاه کروموزوم 17 در توسعه و پیشرفت تومور در NF1 نقش دارند. امروزه مشخص شده است که ژن نوروفیبروماتین در ایجاد تومورهایی غیر از نوروفیبروماتوز تیز نقش دارد که از جمله آنها میتوان به کارسینومای کولون، نوروبلاستوما و ملانومای بدخیم اشاره کرد. این مشاهدات تائید میکنند که نوروفیبروماتین نقش مهمی در رشد و تمایز سلولی بر عهده دارد.
همبستگی ژنوتیپ- فنوتیپ:
در ژن نوروفیبرومین بیش از 100 جهش متفاوت شامل حذفشدگیها، درجیها یا واردشدگیها، دوتاشدگیها و جایگزینیها نقطهای شناسایی شده است. بیشتر این جهشها به ناقصشدگی شدید پروتئین یا فقدان کامل بیان ژنها منجر میشوند. تاکنون شواهد اندکی پیرامون ارتباط بین جهشهای ویژه و علائم بالینی وجود دارد. این امر با گزارشهایی که در رابطه با گوناگونی چشمگیر و قابلتوجه درون خویشاوندی ارائه شده است، سازگاری دارد و بر تأثیرات احتمالی ژنهای تعدیلکننده اشاره میکند. مبتلایان واجد حذفشدگیهای بزرگ مانند حذف همه ژن NF1، شدت ابتلای بهمراتب بیشتری دارند که از جمله آنها میتوان به مشکلات یادگیری و ذهنی، قامت مارفانوئیدی و تعداد بسیار زیاد نوروفیبروما اشاره کرد.
نوروفیبروماتوز تیپ 2
در بیماری NF2 هم لکههای شیرقهوهای و هم نوروفیبروماتا رخ میدهد، اگرچه نسبت به NF1 شیوع بسیار کمتری دارند. شاخصترین نشانه NF2 بروز تومورها در اوایل دوران بلوغ است که عصب هشتم مغزی را گرفتار میکنند. به این تومورها گاهی هنوز نرومای آکوستیک گفته میشود ولی امروزه بجای آن واژهی شوانوماهای وستیبولار ترجیح داده میشود. انواعی از تومورهای دیگر سیستم عصبی مرکزی نیز به فراوانی رخ میدهند، هرچند بیش از نیمی از این موارد بدون نشانه هستند. در مواردی شوانوماهای نخاعی و محیطی نیز ایجاد میشود که این حالات را شوانوماتوز مینامند. یکی از علائمی که فقط در NF2 وجود دارد و در NF1 دیده نمیشود کاتاراکت است، البته این عارضه خفیف بوده و نیازی به درمان ندارد.
لوکوس NF2 بر روی کروموزوم 22q وجود دارد. ژن مربوطه، شوانومین، در سال 1993 کلون شد. این ژن گسترهی 110kb داشته و 17 اگزون دارد. تصور میشود فراوردهی این ژن (که گاهی مرلین خوانده میشود) جزئی از اسکلت سلولی است و همچنین بهعنوان فرونشانندهی توموری عمل میکند.
تشخیص
در بیوپسی نوروفیبرومها تجمعات بدون کپسول و با حدود مشخص از رشتههای عصبی کوچک و سلولهای دوکیشکل با هستههای متموج داخل درم مشاهده میشود (شکل 2). نوروفیبرومهای پلکسی فرم در امتداد مسیر اعصاب محیطی به وجود میآیند و باعث ایجاد ندولهای بزرگ میشوند که غالباً روی آنها هیپرپیگمانتاسیون و هیپرتریکوز وجود دارد. این تومورها در 20 درصد افراد دچار نوروفیبروماتوز ایجاد میشود و در 2 درصد موارد دچار استحاله بدخیم میگردند (بیش از 40 سالگی نادر است). تشخیص از راه کلینیکی با پزشک است. MRI و CT در تشخیص قابلاستفاده است.
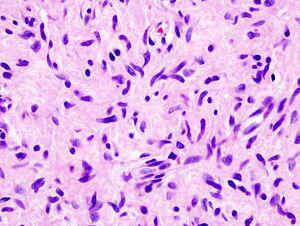
شکل 2: بیوپسی نوروفیبروم رنگآمیزی شده
موتاسیون یا جهشهای جدید (خودبخودی) را نمیتوان از طریق آزمایش ژنتیک مشخص نمود، چراکه قابل پیشبینی نیست. در مورد زوجهای درگیر این بیماری میتوان ابتدا با انجام تست مربوطه نوع جهش بیماریزا را تعیین کرد و در هفته دهم بارداری با نمونهبرداری از طریق آمنیوسنتز amniocentesis یا نمونهبرداری از ویلوس کوریونیک chorionic villus جنین، تشخیص قبل از تولد انجام پذیرد.
درمان
درمانی قطعی برای این بیماری وجود ندارد ولی در صورت لزوم (بخصوص به دلایل زیبایی یا فشار بر نواحی حساس) جراحی و برداشتن برخی غدهها در بعضی افراد میتواند مؤثر باشد، اما ضایعات ممکن است عود بکنند. برای بیماران مشاوره ژنتیکی پیشنهاد شده و توصیه میشود برای بررسی بالینی هر 6 ماه یکبار جهت نظارت بر ندولها و هر شرایطی که با نوروفیبروماتوزیس همراهی دارد مراجعه نمایند. در نوروفیبروماتوز نوع دو، با استفاده از تکنولوژیهای پیشرفتهی تشخیصی میتوان تومورهای کوچک حتی با ابعاد میلیمتری را شناسایی نمود و به این صورت درمان زودهنگام را آغاز نمود.
برداشتن تومورها به طور کامل از طریق جراحی انتخابی بوده و در صورت درگیری در برخی موارد ممکن است موجب از دست دادن شنوایی در بیمار شود. سایر روشها در درمان نوروفیبروماتوز شامل برداشتن نسبی تومور و سپس رادیوتراپی یا شیمیدرمانی است. اگر چنانچه رشد تومورها سریع نباشد بهتر آن است که بیمار را فقط تحتنظر قرار داد.
کاربردهای بالینی و چشماندازهای آینده
مشخص شدن نقشه ژن نوروفیبرومین، توانایی انجام تشخیص پیش از تولد و تشخیص پیش از بروز علائم را با استفاده از آنالیز پیوستگی یا بررسی مستقیم جهش فراهم نموده است. در عمل تعداد بسیار کمی از خانوادهها مایل به پیگیری و استفاده از راهکارها هستند. بخشی از این امر به دلیل آن است که NF1 را بهعنوان یک بیماری خطرناک نمیشناسند و قسمتی به این دلیل است که آنالیز جهش در پیشبینی شدت بیماری کمککننده نیست. در حال حاضر درمانی برای NF1 وجود ندارد.
در غیاب ژندرمانی، استفاده از دارودرمانی در جهت تنظیم افزایشی فعالیت GAP نوروفیبرومین یا تنظیم کاهشی فعالیت RAS میتواند مفید واقع شود، بههرحال مشکل آن است که چگونه این راهکارها در مورد بافتهای گوناگون هدف مانند سیستم عصبی مرکزی به کار گرفته شود. در مجموع شناسایی و کلون کردن ژن بیماریهای NF1 و NF2 تنها منجر به شناخت بهتر مسیرهای بیماریزایی شده است و امید آن میرود در آینده با این گونه مطالعات بتوان راهکارهای درمانی تازهای معرفی کرد.
https://www.mayoclinic.org/diseases-conditions/neurofibromatosis/symptoms-causes/syc-20350490
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام