ارتباط ویروس ایدز (HIV) با بیماریهای قلبی – عروقی
فاطمه شاهد1 – اصغر الهی2
1: کارشناسی علوم آزمایشگاهی علوم پزشکی قم
2: عضو هیئت علمی علوم پزشکی قم
مقدمه
ویروس HIV یکی از اعضای خانوادهی لنتیویروسها و عامل ایجاد بیماری ایدز است. ایدز با سرکوب شدید ایمنی، عفونتهای فرصتطلب، تومورهای بدخیم، لاغری مفرط و تخریب سیستم عصبی مرکزی همراه است. ویروس HIV انواع مختلفی از سلولهای سیستم ایمنی شامل سلولهای T کمکی +CD4، ماکروفاژها و سلولهای دندریتیک را آلوده میکند (1). در افراد +HIV برای درمان ویروسی از ترکیبات ضد رتروویروسی (CART) استفاده میشود که این ترکیبات HIV را بهعنوان یک بیماری مزمن کنترل میکنند. در این افراد به علت اینکه ایمنی بهطور مداوم فعال است، باعث بروز اختلالات پاتوفیزیولوژیک در عفونت اولیه میشود که منجر به تعویض مسیر متابولیک سلولهای ایمنی و فعال شدن مسیرهای انعقادی میگردد.
HIV، التهاب و بیماریهای قلبی- عروقی (CVD)
بیماریهای قلبی عروقی بر اثر عواملی همچون تغذیهی نامناسب، استرس روزمره، چربی خون بالا و … ایجاد میشوند. جالب است بدانید آلودگی با ویروس HIV نیز یکی از علل ابتلا به بیماریهای قلبی عروقی همچون آترواسکلروزیس است. مطالعات تاکنون نشان میدهند که ابتلا به CVD در افراد +HIV یک فرآیند پیچیده است که چندین پارامتر را شامل میشود، ازجمله فاکتورهای خطر سنتی مثل سیگار کشیدن، عوارض جانبی CART (بخصوص مهارکنندههای پروتئاز HIV) و همچنین فعالسازی ایمنی مزمن و تغییرات متابولیکی مرتبط با آن که مهمترین عامل CVD است (2،3،4).
با گسترش CART عوارض آن مانند افزایش فشارخون ریوی و مرگومیر ناگهانی قلبی بهطور شایع در افراد +HIV افزایش مییابد، بهطوریکه انفارکتوس میوکارد قلب در افراد +HIV دو برابر افراد غیرآلوده است (5)، بنابراین اگرچه CART زندگی افراد آلوده را طولانیتر میکند اما خطر ابتلا به بیماریهای غیرایدز مثل CVD را افزایش میدهد (6) بهطوریکه یک علت مهم مرگومیر در افراد HIV+،CVD است (7).
نقش منوسیتها و ماکروفاژها
ماکروفاژها از منوسیتها مشتق میشوند و سلولهای ارائهدهندهی آنتیژن مهمی هستند که در بیماریزایی HIV نقش محوری دارند (8). منوسیتها توانایی پاسخ به محرکهای محیطی متفاوت مثل سایتوکاینها را دارند و پاسخدهندههای اصلی به LPSهای (لیپوپلیساکارید) افزایشیافته به علت انتقال میکروبی هستند (9)، بنابراین آنها راههای پیش التهابی راه میاندازند که در این راهها سایتوکاینهای قدرتمندی مثل IL-6 و TNF-a را تولید میکنند و این سایتوکاینها التهاب موضعی و سیستمیک را آغاز میکنند (10، 11).
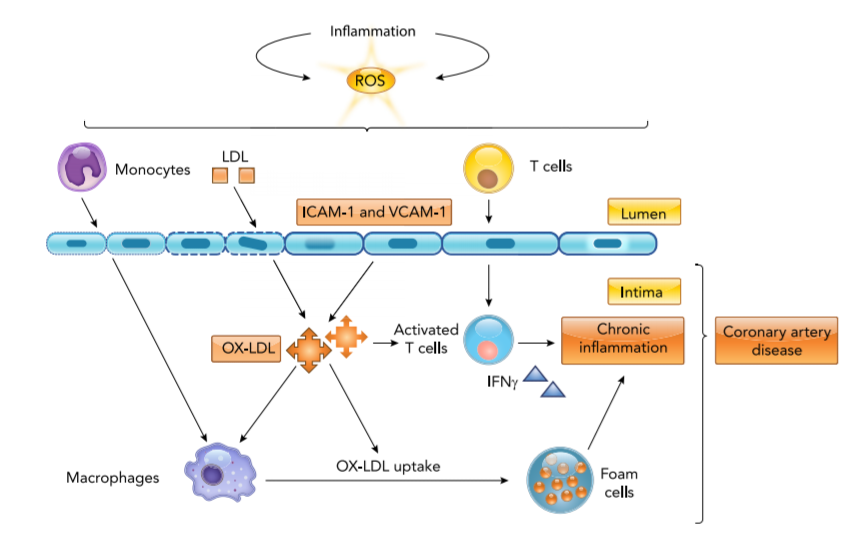
تصویر 1: فعالسازی ایمنی، استرس اکسیداتیو و شروع آترواسکلروزیس
التهاب وابسته به HIV و پروتئینهای ویروسی مثل Tat، Npr،Nef و gp120 میتوانند گونههای اکسیژن فعال (ROS) تولید کنند و منجر به افزایش استرس اکسیداتیو شوند (12). این گونههای اکسیژن فعال،LDL را اکسید میکنند و منجر به تشکیل OX-LDL در درونیترین غشاء پوششی دیوارههای شـــــــریان (Intima) میشوند. OX-LDL و سایتوکاینهای التهابی، بیان ملکولهای چسبندگی (ICAM-1 و VCAM-1) را روی اندوتلیال عروق و سلولهای ایمنی افزایش میدهند (13). چنین ملکولهای چسبندگی، مهاجرت سلولهای T به Intima را افزایش میدهند؛ سلولهای T توسط OX-LDL و سایتوکاینهای تولیدشده مثل INF فعال میشوند و به افزایش التهاب و بیماری آترواسکلروزیس کمک میکنند (14). OX-LDL همچنین میتواند ماکروفاژها را فعال کند. منوسیتهای فعالشده پس از انتقال از اندوتلیال به ماکروفاژ تمایز مییابند و میتوانند مقدار بیشتری OX-LDL را جذب کنند؛ سرانجام به سلولهای پر از چربی به نام سلولهای فوم تبدیل میشوند و پلاکهای اولیهی چربی را تشکیل میدهند. با بیان IL-4 و CD36 روی ماکروفاژها، جذب OX-LDL و تشکیل سلولهای فوم افزایش مییابد (15). همهی این وقایع ریسک ابتلا به بیماری شریان کرونری را در افراد +HIV افزایش میدهند (تصویر 1). همچنین DCها (سلولهای دندریتیک) با آنتیژنهای آترواسکلروزیس برخورد میکنند و برای ارائه آنتیژن و ترویج آنتیژن خاص بیماریزا یا فعال کردن سلولهای T تنظیمی به ارگانهای لنفوئیدی ثانویه مهاجرت میکنند (16).
تغییرات در زیرمجموعههای منوسیت در طول مراحل مختلف آلودگی به HIV نقش مهمی در بیماریزایی HIV دارد (17). منوسیت سه زیرشاخه دارد که شامل منوسیت غیرکلاسیک، منوسیت متوسط و کلاسیک است. منوسیتهای غیرکلاسیک و متوسط در افراد +HIV افزایش مییابند و سطوح بالای رسپتورهای کموکاین (CCR2 و CCR5) و رسپتور فراکتالکین (CX3CR1) را بیان میکنند، درحالیکه منوسیت کلاسیک در فاگوسیتوز نقش دارد و آنتیباکتریال است (11). منوسیت متوسط همچنین بهعلت سطوح نسبتاً بالای سایتوکاینهای پیشالتهابی که ترشح میکند، بهطور مستقل در CVD نقش دارد (18).
نقش سلولهای T
در عفونت حاد HIV و ارائهی اولیهی ویروس، فعالسازی پاسخ ایمنی تطبیقی زودهنگام اتفاق میافتد (19). با ورود ویروس به بدن، ذرات آن توسط DCها گرفته میشود. این سلولها الگوهای ملکولی پاتوژن را از طریق toll-like receptorها شناسایی میکنند، سپس پاتوژنها را به قطعات پروتئینی که اپیتوپ نامیده میشوند میشکنند. اپیتوپها توسط MHC I به +TCD8 و توسط MCH II به +CD4 ارائه میشوند و درنهایت +CD4 و +CD8 فعال میشوند (20)، سپس تکثیر مییابند و زیرمجموعههای مربوطه شروع به ترشح کموکاین و سایتوکاین میکنند. پاسخ سلولهای TH(T کمکی) به توسعهی آترواسکلروزیس کمک میکند درحالیکه سلولهای Tتنظیمی مانع تکثیر سلولهای T اجرایی و توسعهی آترواسکلروزیس میشوند که این عدم تعادل بین TH پاتوژنیک و Tتنظیمی منجر به توسعهی آسیب میشــــود. سطوح بالاتر ROS همچنین میتواند TGF-β را فعال کند که میتواند سلولهای T تنظیمی را در عفونت HIV بوجود بیاورد و به عدم تعادل با سلولهای TH کمک کند (21)، بنابراین سلولهای T تنظیمی یک نقش مهم در جلوگیری از آترواسکلروزیس و همچنین در کنترل التهاب در عفونت HIV دارند.
فعال شدن مکرر سلولهای T در عفونت HIV منجر به کاهش ظرفیت تکثیر این سلولها میشود (22) که بهنوبهی خود منجر به از دست دادن ظرفیت تولید سایتوکاین و بیان مارکرهای فنوتایپ خستگی (مرگ سلولی برنامهریزیشده) و بیان پایینتر ملکولهای تحریککننده CD28 میشود و چون CD28 در تنظیم پاسخهای ایمنی و حفظ تحمل محیطی نقش مهمی دارد (23)؛ افزایش تعداد +TCD8 هایی که CD28 منفیاند میتواند منجر به CVD سابکلینیکال شودکه با عوارض و مرگومیر بالا مرتبط است (4، 24).
دو فرآیند اصلی سلولی مسئول متابولیسم گلوکز، فسفریلاسیون اکسیداتیو و گلیکولیز هستند که فسفریلاسیون اکسیداتیو برای تولید انرژی زیاد است و تولیدات واسطهی گلیکولیز برای سنتز نیازهای تکثیر و تمایز سلولهای ایمنی ضروری هستند. برای اینکه یک سلول از حالت غیرفعال به حالت فعال تبدیل شود، نیازمند تعویض مسیر متابولیک خود است. در عفونت HIV، منوسیتها و سلولهای T خاموش، فعال میشوند و بیان Glut-1 روی غشاء پلاسمایی آنها افزایش مییابد و فنوتایپ متابولیکشان به گلیکولیتیک تغییر میکند (25) که این تعویض برای تمایز منوسیت به ماکروفاژ ضروری است. دادههای موجود نشان میدهد که تغییرات متابولیک (اغلب افزایش گلیکولیز) در سلولهای ایمنی با استرس اکسیداتیو بیشتری همراه است و یک محیط پیشالتهابی ایجاد میکند که خطر ابتلا به CVD را در افراد +HIV افزایش میدهد. همچنین بیان بیش از اندازهی Glut-1 میتواند تولید سایتوکاینهای پیشالتهابی مثل IL-6 را از طریق مکانیسم وابسته به ROS افزایش دهد. علاوه بر این، مصرف بالای گلوکز بیان سایتوکاینهای پیشالتهابی مثل TNF-α و IL-1β را افزایش میدهد که هردو به همانندسازی HIV کمک میکنند. گلیکولیز همچنین مسیر پنتوز فسفات را برای تولید پیشسازهای موردنیاز سنتز پروتئین و نوکلئوتید تحریک میکند.
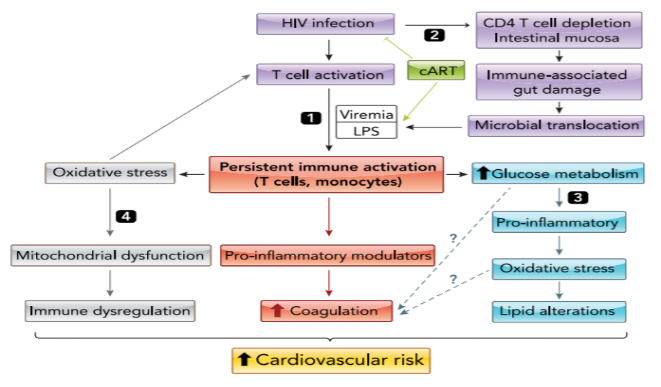
تصویر 2: نقش اختلالات ایمنومتابولیک در ابتلا به CVD در افراد +HIV
- سلولهای Tبا عفونت HIV فعال میشوند اما ویرمی با سطح پایین و LPS القاشده توسط CART باعث فعالسازی ایمنی مزمن میشوند که درنهایت سایتوکاین تولید میشود و انعقاد افزایش مییابد.
- در ابتدای عفونت HIV، کاهش قابلتوجه +CD4 در موکوس روده اتفاق میافتد، سپس اپیتلیال روده آسیب میبیند و LPS که جزو اصلی دیواره سلولی باکتریهای گرم منفی است از روده به خون و بافت ترشح میشود که بیشتر، باعث فعالسازی ایمنی مزمن میشود و در ادامهی آن استرس اکسیداتیو افزایش مییابد و یک چرخه تشکیل میشود که بیشتر، سلولهای T را فعال میکند.
- افزایش بیان Glut-1 باعث افزایش متابولیسم گلوکز در لنفوسیتهای Tمیشود که باعث التهاب و استرس اکسیداتیو میگردد که بهنوبهی خود منجر به تغییرات چربی مانند افزایش OX-LDL میشود. احتمالاً افزایش متابولیسم گلوکز و استرس اکسیداتیو با افزایش انعقاد مرتبطاند.
- افزایش استرس اکسیداتیو سبب آسیب میتوکندری در لنفوسیتهای Tمیشود که باعث اختلال در تنفس و تولید ATP در میتوکندری میشود.
درنتیجهی این موارد، خطر ابتلا به CVD در افراد +HIV افزایش مییابد (تصویر 2).
REFERENCES:
- K abbas. Cellular and molecular immunology. 2015
- Baker JV, Lundgren JD. Cardiovascular implications from untreated human immunodeficiency virus infection. Eur Heart J 32: 945–951, 2011. doi:10.1093/eurheartj/ehq483.
- Dixon DL, Griggs KM, Bersten AD, De Pasquale CG. Systemic inflammation and cell activation reflects morbidity in chronic heart failure. Cytokine 56: 593–599, 2011. doi:10.1016/j.cyto.2011.08. 029.
- Kaplan RC, Sinclair E, Landay AL, Lurain N, Sharrett AR, Gange SJ, Xue X, Hunt P, Karim R, Kern DM, Hodis HN, Deeks SG. T cell activation and senescence predict subclinical carotid artery disease in HIV-infected women. J Infect Dis 203: 452–463, 2011. doi:10.1093/infdis/jiq071.
- Triant VA. HIV infection and coronary heart disease: an intersection of epidemics. J Infect Dis 205, Suppl 3: S355–S361, 2012. doi:10.1093/ infdis/jis195.
- Serrano-Villar S, Gutiérrez F, Miralles C, Berenguer J, Rivero A, Martínez E, Moreno S. Human immunodeficiency virus as a chronic disease: evaluation and management of nonacquired immune deficiency syndrome-defining conditions. Open Forum Infect Dis 3: ofw097, 2016. doi:10. 1093/ofid/ofw097.
- TsengZH,SecemskyEA,DowdyD,VittinghoffE, Moyers B, Wong JK, Havlir DV, Hsue PY. Sudden cardiac death in patients with human immunodeficiency virus infection. J Am Coll Cardiol 59: 1891–1896, 2012. doi:10.1016/j.jacc.2012.02. 024.
- Coleman CM, Wu L. HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6: 51, 2009. doi:10.1186/ 1742-4690-6-51.
- Funderburg NT, Mayne E, Sieg SF, Asaad R, Jiang W, Kalinowska M, Luciano AA, Stevens W, Rodriguez B, Brenchley JM, Douek DC, Lederman MM. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood 115: 161–167, 2010. doi:10. 1182/blood-2009-03-210179.
- Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenicandregulatorypathways.PhysiolRev 86: 515–581, 2006. doi:10.1152/physrev.00024. 2005.
- Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2: 1, 2014. doi:10. 1186/2050-7771-2-1.
- PorterKM,SutliffRL.HIV-1,reactiveoxygenspecies, and vascular complications. Free Radic Biol Med 53: 143–159, 2012. doi:10.1016/j. freeradbiomed.2012.03.019.
- Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 21: 1876– 1890, 2001. doi:10.1161/hq1201.100220.
- Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis.ClinImmunol134:33–46,2010.doi:10.1016/ j.clim.2009.07.002.
- Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355, 2011. doi:10.1016/j.cell.2011.04.005.
- Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res 50, Suppl: S364–S369, 2009. doi: 10.1194/jlr.R800092-JLR200.
- ZawadaAM,RogacevKS,SchirmerSH,SesterM, Böhm M, Fliser D, Heine GH. Monocyte heterogeneity in human cardiovascular disease. Immunobiology 217: 1273–1284, 2012. doi:10.1016/j. imbio.2012.07.001.
- Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, Große-Dunker G, Heisel I, Hornof F, Jeken J, Rebling NM, Ulrich C, Scheller B, Böhm M, Fliser D, Heine GH. CD14CD16monocytes independently predict cardiovascular events: a cohort study of 951 patients referred forelectivecoronaryangiography.JAmCollCardiol 60: 1512–1520, 2012. doi:10.1016/j.jacc. 2012.07.019.
- McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol 10: 11– 23, 2010. doi:10.1038/nri2674
- Gasper DJ, Tejera MM, Suresh M. CD4 T-cell memory generation and maintenance. Crit Rev Immunol 34: 121–146, 2014. doi:10.1615/ CritRevImmunol.2014010373.
- Amarnath S, Dong L, Li J, Wu Y, Chen W. Endogenous TGF-beta activation by reactive oxygen species is key to Foxp3 induction in TCR-stimulated and HIV-1-infected human CD4CD25- T cells. Retrovirology 4: 57, 2007. doi:10. 1186/1742-4690-4-57.
- Hazenberg MD, Stuart JW, Otto SA, Borleffs JC, Boucher CA, de Boer RJ, Miedema F, Hamann D. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 95: 249–255, 2000.
- Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev 241: 180–205, 2011. doi:10.1111/j.1600-065X.2011.01011.x.
- Shahbaz S, Manicardi M, Guaraldi G, Raggi P. Cardiovascular disease in human immunodeficiencyvirusinfectedpatients:atrueorperceived risk? World J Cardiol 7: 633–644, 2015. doi:10. 4330/wjc.v7.i10.633.
- Palmer CS, Cherry CL, Sada-Ovalle I, Singh A, Crowe SM. Glucose metabolism in T cells and monocytes: new perspectives in HIV pathogenesis. EBioMedicine 6: 31–41, 2016. doi:10.1016/j. ebiom.2016.02.012.
سندرم نقص ایمنی اکتسابی (ایدز)
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام