سندرم سِنسِنبرِنر
Sensenbrenner Syndrome

شاهین اسعدی (دانشجوی دکتری تخصصی ژنتیک پزشکی)
کلیاتی از سندرم سِنسِنبرِنر
سندرم سِنسِنبرِنر که تحت عنوان دیسپلازی کرانیواِکتودرمال نیز شناخته میشود، یک اختلال ژنتیکی است که بر بسیاری از سیستمهای بدن تأثیر میگذارد. شایعترین ویژگیهای این سندرم ناهنجاریهای استخوانی و توسعه غیرطبیعی بافتهای خاصی است که به عنوان بافتهای اِکتودرمال شناخته میشوند و شامل پوست، مو، ناخن و دندان میباشند. علائم و نشانههای این سندرم در بین افراد مبتلا حتی در میان اعضای خانواده، نیز متفاوت است.

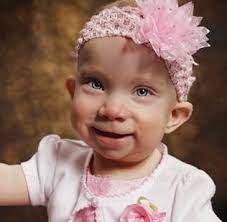
شکل 1: تصویر کودک مبتلا به سندرم سِسِنبرِنر همراه با ویژگیهای متمایز صورت
علائم و نشانههای بالینی سندرم سِنسِنبرِنر
در افراد مبتلا به این سندرم، اختلالات مشخصی از جمجمه و صورت رایج است. اکثر افراد مبتلا به سندرم سِنسِنبرِنر دارای پیشانی برجسته و سر باریک و طولانی شکل (دولیکوسفالی) هستند. انواع مختلف اختلالات صورت میتواند در افرادی که مبتلا به این سندرم هستند، رخ دهد که شامل موارد زیر است:
پایین بودن موقعیت گوشها که ممکن است به سمت عقب چرخیده باشند، فاصله زیاد در گوشههای داخلی چشم (تِلِکانتوس) و گوشههای خارجی چشم که به سمت بالا یا پایین قرار گرفتهاند.

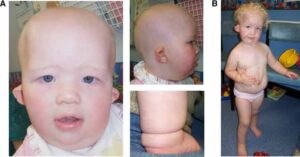
شکل 2: تصاویری از مبتلایان سندرم سِنسِنبرِنر همراه با اختلالات مربوطه
ناهنجاریهای استخوانی مانند اختلال در استخوانهای بلند بازوها و پاها (دیسپلازی متافازی) که منجر به اندام کوتاه و قد کوتاه میشود نیز از علائم این سندرم است. علاوه بر این، افراد مبتلا به سندرم سِنسِنبرِنر اغلب انگشتان کوتاه (براکی داکتیلی) دارند و همچنین برخی از این مبتلایان دارای استخوانهای کوتاه ران و قفسه سینه محدود هستند که میتواند باعث مشکلات تنفسی، بهویژه در نوزادان آسیبدیده شود. لازم به ذکر است که توسعه غیرطبیعی بافتهای اِکتودرمال در افراد مبتلا به سندرم سِنسِنبرِنر میتواند منجر به موهای کمرنگ، دندانهای کوچک و یا مفقود، ناخنهای کوتاه دستها و پاها و شُلی پوست گردد.

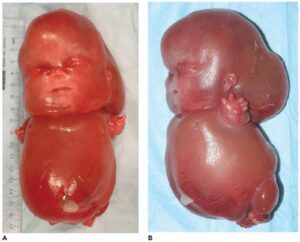
شکل 3: تصاویری از جنینهای مبتلا به سندرم سِنسِنبرِنر همراه با دیسپلازی متافازی
سندرم سِنسِنبرِنر میتواند اعضای بدن و بافتهای دیگر بدن را تحت تأثیر قرار دهد. اختلال کلیوی تحت عنوان نفرونوفتیز در بسیاری از افراد مبتلا به این سندرم رخ میدهد و میتواند منجر به مرگ و میر ناشی از نارسایی کلیه شود که به عنوان بیماری کلیوی در مرحله پایانی (ESRD) شناخته میشود. بعلاوه، اختلالات کبد، قلب و یا چشم نیز در افراد مبتلا به سندرم سِنسِنبرِنر ممکن است رخ دهد.

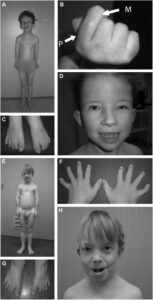
شکل 4: نمایی دیگر از اختلالات مربوطه در سندرم سِنسِنبرِنر
علتشناسی سندرم سِنسِنبرِنر
سندرم سِنسِنبرِنر در اثر جهش حداقل یکی از 4 ژن ایجاد میشود که عبارتند از: ژن WDR35 که در بازوی کوتاه کروموزوم شماره 2 بهصورت 2p24.1 مستقر است. ژن IFT122 که در بازوی بلند کروموزوم شماره 3 بهصورت 3q21.3-q22.1 مستقر است. ژن WDR19 که در بازوی کوتاه کروموزوم شماره 4 بهصورت 4p14 مستقر است و ژن IFT43 که در بازوی بلند کروموزوم شماره 14 بهصورت 14q24.3 مستقر است. پروتئین تولیدشده از هر یک از این ژنها تحت عنوان کمپلکس IFT-A شناخته میشود. این کمپلکس در ساختارهای شبیه انگشت به نام مژهها یافت میشود که از سطح سلولها خارج میشوند. این ساختارها برای توسعه و عملکرد بسیاری از انواع سلولها و بافتها مهم هستند. کمپلکس IFT-A در فرآیندی به نام حملونقل داخلی فلاژلار دخیل است که مواد را با مژهها حرکت میدهد. این حرکت مواد برای جمعآوری و نگهداری این ساختارها ضرورت دارد. کمپلکس IFT-A مواد را از رأس تا مرکز مژهها حمل میکند.

شکل 5: نمای شماتیک از کروموزوم شماره 2 که ژن WDR35 در بازوی کوتاه این کروموزوم بهصورت 2p24.1 مستقر است
جهش در هر یک از این چهار ژن ذکرشده، مقدار یا عملکرد یکی از زیرواحدهای IFT-A را کاهش میدهد. کمبود یا عملکرد غیرطبیعی یکی از زیرواحدهای کمپلکس IFT-A، عملکرد کل این کمپلکس را مختل میکند که مانع از مونتاژ و نگهداری مژههای سطح سلولی میشود. این جهشها منجر به کاهش تعداد مژههای سطح سلولی و ایجاد ناهنجاری در شکل و ساختار آنها میشود. شایان ذکر است که حدود 40 درصد از افراد مبتلا به سندرم سِنسِنبرِنر جهش در یکی از چهار ژن ذکرشده را دارند.

شکل 6: نمای شماتیک از کروموزوم شماره 3 که ژن IFT122 در بازوی بلند این کروموزوم بهصورت 3q21.3-q22.1 مستقر است
سندرم سِنسِنبرِنر از الگوی توارثی اتوزومال مغلوب پیروی میکند؛ بنابراین برای ایجاد این سندرم، دو نسخه از ژنهای جهشیافته WDR35 ,IFT122 ,WDR19 ,IFT43 (یکی از پدر و دیگری از مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به سندرم سِنسِنبرِنر در حالت اتوزومی مغلوب برای هر بارداری احتمالی به میزان 25% است.

شکل 7: نمای شماتیک از کروموزوم شماره 4 که ژن WDR19 در بازوی کوتاه این کروموزوم بهصورت 4p14 مستقر است
فراوانی سندرم سِنسِنبرِنر
سندرم سِنسِنبرِنر اختلال ژنتیکی نادر است که فرکانس شیوع آن در جهان مشخص نیست. تاکنون حداقل 40 مورد مبتلا به این سندرم از سراسر جهان در ادبیات پزشکی گزارش شده است.

شکل 8: نمای شماتیک از کروموزوم شماره 14 که ژن IFT43 در بازوی بلند این کروموزوم بهصورت 14q24.3 مستقر است
تشخیص سندرم سِنسِنبرِنر
سندرم سِنسِنبرِنر بر اساس یافتههای بالینی و فیزیکی مبتلایان و برخی آزمایشهای پاتولوژیکی، تشخیص داده میشود. دقیقترین روش تشخیص این سندرم، آزمایش ژنتیک مولکولی برای ژنهای ذکرشده بهمنظور بررسی وجود جهشهای احتمالی است. تشخیص پیش از تولد نیز با استفاده از تکنیک PGD و مایع آمنیوسنتز و یا نمونهبرداری از پرزهای کوریونی جفت جنین امکانپذیر است.
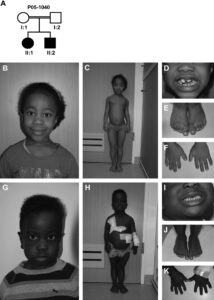
شکل 9: نمای کلی از اختلالات سندرم سِنسِنبرِنر
مسیرهای درمانی سندرم سِنسِنبرِنر
استراتژی درمان و مدیریت سندرم سِنسِنبرِنر بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص مغز و اعصاب، متخصص پوست، متخصص ریه، نفرولوژیست، متخصص چشم، جراحان و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان قاطعی برای این سندرم وجود ندارد و تمامی اقدامات بالینی بهمنظور تخفیف رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که خواستار فرزندی سالم هستند، از جایگاه ویژهای برخوردار است.
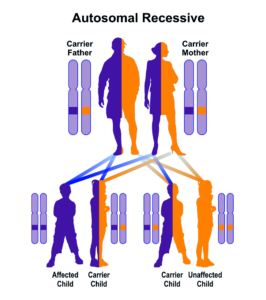
شکل 10: نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم سِنسِنبرِنر از این الگو تبعیت میکند
تاریخچه سندرم سِنسِنبرِنر
سندرم سِنسِنبرِنر برای اولین بار در سال 1975 توسط Sensenbrenner JA و همکاران گزارش گردید.
منبع:
دکتر اسعدی شاهین، دکتر ملتیار حسن، دکتر روشنروان ندا، دکتر دادشپور مهدی، جمالی مهسا، محمدزاده حمیده، فتاحی محیا، کتاب پاتولوژی در ژنتیک پزشکی جلد 6 (M-Y)، صفحات 762-753، انتشارات کتب دانشگاهی عمیدی، بهار 1397.
سندرم بال پروانهای Epidermolysis Bullosa (EB) Syndrome
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام