سندرم باردت بیدل
Bardet-Biedl Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، دکتر روشنک سامبرانی (متخصص ژنتیک مولکولی)، سپیده باستانی (کارشناس ارشد بیوشیمی)، وقار اکبرپور (دانشجوی کارشناسی ارشد ژنتیک)

نگارنده مسئول: شاهین اسعدی (Molecular Geneticist)
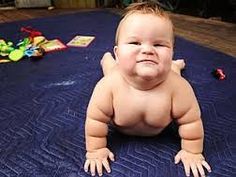
سندرم باردت بیدل، یک اختلال ژنتیکی کلیوپاتیک انسانی است که در بسیاری از سیستمهای بدن تأثیر میگذارد. این اختلال عمدتاً با چاقی، رتینیت پیگمانتوزا، پلیداکتیلی، هیپوگنادیسم و در برخی موارد نارسایی کلیه مشخص میشود.
این سندرم در درجه اول با اختلال در سلولهای محرک نور (سلولهای مخروطی و میلهای) شبکیه چشم بهصورت دیستروفی کانراد سلولها مشخص میشود.
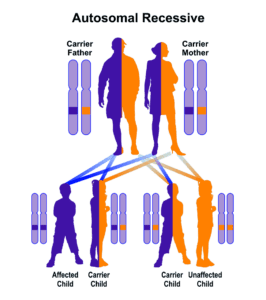
همچنین در این بیماری، چربی بهطور نامتناسب در ناحیه شکم و سینه تجمع یافته و مشکلاتی در یادگیری شخص مبتلا ایجاد میگردد. اختلالات بصری با مرور زمان، ممکن است بدتر شود و منجر به کوری شخص مبتلا به سندرم باردت بیدل گردد. با از دست دادن حس بینایی، مشکلات یادگیری افزایش مییابد و در برخی موارد منجر به اختلال شدید روانی در این افراد میشود. سندرم باردت بیدل از الگوی توارثی اتوزومال مغلوب پیروی میکند. سندرم باردت بیدل همپوشانی قابلتوجهی با یک اختلال ژنتیکی دیگر بنام سندرم لورنسمون نشان میدهد؛ در واقع در گذشته این اختلالات، باهم یک نوع اختلال در نظر گرفته میشد بنام سندرم لورنس باردت بیدل اما با پیشرفت زیستشناسی مولکولی این دو اختلال از هم تفکیک شدند و بهصورت جداگانه، ارجاع داده شدند.
بااینحال تحقیقات اخیر نشان داده است که برخی از افراد با یافتههای بالینی سندرم لورنس موون، جهش در ژن مرتبط با این سندرم را داشتهاند، بنابراین این کشف باعث شده است تا ژنتیکدانان این دو اختلال را بهعنوان یک اختلال در نظر بگیرند.
علائم و نشانهها
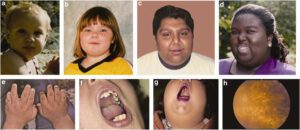
علائم و نشانههای سندرم باردت بیدل حتی در میان افراد درون یک خانواده تا حد زیادی متفاوت است، علاوه بر این، شدت علائم خاص نیز ممکن است تا حد زیادی متفاوت باشد. از ویژگیهای اصلی این سندرم میتوان به اختلال دیستروفی کانراد در سلولهای شبکیه چشم، پلیداکتیلی در دست و پا، چاقی مفرط، اختلالات کلیه و مشکلات یادگیری اشاره کرد.
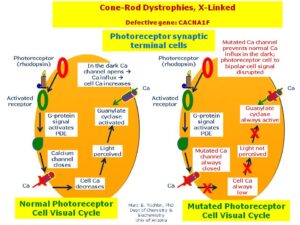
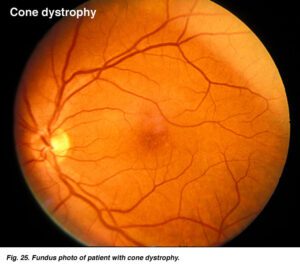
دیستروفی کانراد شکلی از اختلال در عملکرد شبکیه چشم است. شبکیه دارای غشاء حساس به نور است که بر اساس آن تصاویر در پشت چشم متمرکز میشود. سلولها در شبکیه چشم به شکل مخروطی یا میلهای هستند که بهعنوان گیرنده عمل میکنند و نور دریافتی را به تکانههای عصبی ارسال و تکانههای عصبی این نور دریافتی را به تصویر موردنظر، ترجمه میکنند که در سندرم باردت بیدل این تکانههای عصبی بهتدریج، زوال مییابند (دیستروفی کانراد) و باعث از دست دادن حس بینایی در شخص مبتلا میشود. نشانههای دیستروفی کانراد، ممکن است 7 تا 8 سال پس از ابتلای شخص به سندرم باردت بیدل، آشکار گردد که در پی آن شخص مبتلا به سندرم باردت بیدل، اختلال دید در نور کم یا عدم توانایی بینایی در مناطق تاریک را خواهد داشت و بهتدریج شخص مبتلا بینایی خود را از دست خواهد داد.
پیشرفت و درجه اختلال بینایی در میان افراد مبتلا، متفاوت است؛ در برخی موارد انحطاط شبکیه ممکن است دوره مشخصه رتینیت پیگمانتوزا را دنبال کند که همراه با شبکوری و یا از دست دادن توانایی تشخیص رنگ خواهد بود.
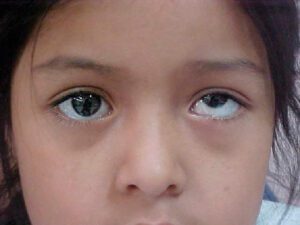
در این سندرم ممکن است علائم دیگری مانند لوچی چشم (استرابیسموس)، حرکت سریع چشم بهصورت غیرارادی و غیرطبیعی (نیستاگموس)، کدر شدن عدسی چشم (آبمروارید) و آسیب به عصب بینایی که اغلب با افزایش فشار درون چشم (گلوکوم) همراه است، مشاهده گردد.

در حدود 70% از افراد مبتلا به این سندرم ، پلیداکتیلی یا حضور انگشتان اضافی در دستها در نزدیکی انگشت کوچک و در پاها در نزدیکی انگشت پنجم، مشاهده میشود و همچنین چسبندگی انگشتی میان انگشتان دست و پا، ممکن است رخ دهد، بعلاوه ممکن است انگشتان دست و پا بهصورت کوتاه با حالت قوس و یا گسترده و صاف و یا حلقهوار شدن انگشت کوچک دست نیز دیده شود.
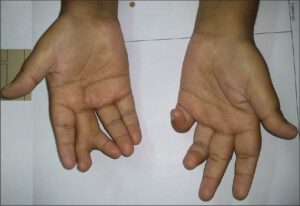
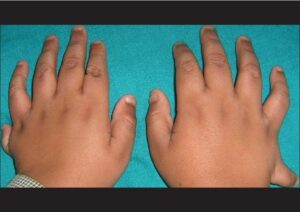
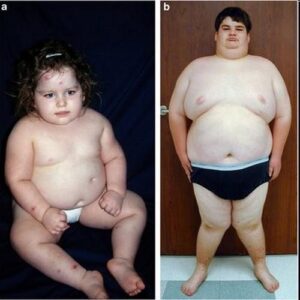
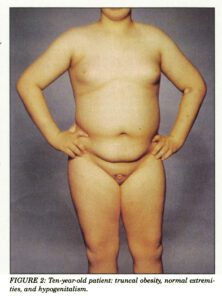
یکی دیگر از علائم رایج در این سندرم ، توزیع نامناسب چربی بدن در ناحیه شکم و سینه است که منجر به چاقی مفرط میشود. افراد مبتلا به سندرم باردت بیدل در هنگام تولد، وزنشان طبیعی است اما افزایش وزن در سال اول زندگی آشکار میشود.
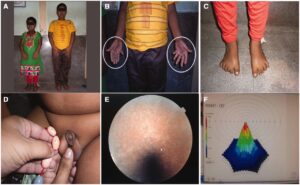
مشکلات یادگیری خفیف تا متوسط در افراد مبتلا به سندرم باردت بیدل رایج است. در گزارشهای اولیه در این سندرم، ناتوانیهای یادگیری به عقبماندگی ذهنی نسبت داده شد، اما مطالعات اخیر نشان داده است که تنها تعداد محدودی از مبتلایان این سندرم ، دارای ضریب هوشی پایین قابلتوجهی هستند که این افراد در مشکلات یادگیری نیز بسیار قابلتوجه میباشند. افراد مبتلا به سندرم باردت بیدل، ممکن است تأخیر در رسیدن به نقاط عطف رشد را تجربه کنند.
.
سندرم باردت بیدل با کاهش عملکرد غدد جنسی بیضه در مردان (هیپوگنادیسم) همراه است. اختلال مازاد در مردان مبتلا به این سندرم شامل آلت تناسلی کوچک، شکسته شدن بیضهها و فرود به داخل کیسه بیضه و تأخیر در رسیدن به سن بلوغ یا عدم بلوغ میباشد. زنان مبتلا به سندرم باردت بیدل ممکن است دارای اختلالات ادراری تناسلی و عدم توسعه لولههای فالوپ (هیپوپلازی) و عدم توسعه رحم و تخمدان باشند و همچنین دوبلکسی رحم، تنگی نسبی یا کامل مهبل واژن (آترزی)، ترشح مجموعهای از مایعات اضافی آبکی در رحم و واژن، گذرگاههای غیرطبیعی بین مثانه و واژن، عدم وجود سوراخ واژن یا سوراخ مجرای ادراری و بینظمیهای قاعدگی در برخی زنان گزارش شده است.
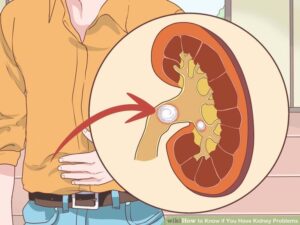
در برخی از افراد مبتلا به سندرم باردت بیدل ممکن است ناهنجاریهای ساختاری یا عملکردی در توسعه کلیهها دیده شود که منجر به تجمع غیرطبیعی ادرار در کلیهها (هیدرونفروز) و یا التهاب کلیه و لگن (پیلونفریت) میگردد. در برخی موارد اختلالات کلیه میتواند بسیار شدید باشد که به دیالیز یا حتی پیوند کلیه منجر شود.

کودکان مبتلا به سندرم باردت بیدل، ممکن است تأخیر در صحبت یا اختلال در سخن گفتن را تجربه کنند. کودکان مبتلا ممکن است صدایی با تن بالا اما بیان ضعیف داشته باشند؛ برای مثال ممکن است این کودکان در تولید آوا برای صحبت با تلفن یا موبایل دچار مشکل شوند و در واقع توانایی تولید صامت جایگزین در آغاز کلمه را نخواهند داشت و حذف صامت در پایان کلمه را خواهند داشت.
اختلال رفتاری در سندرم باردت بیدل نیز گزارششده است؛ افراد مبتلا به این سندرم ممکن است اضطراب، افسردگی، اختلالات خلقی و اختلالات وسواس (OCD) را نشان دهند. برخی از افراد ممکن است علائمی مانند بیماری اوتیسم را نشان دهند.


سندرم باردت بیدل، میتواند با فشارخون بالا، از دست دادن حس بویایی (آبنوسمی)، عدم حفظ تعادل بدن و در نتیجه راه رفتن غیرطبیعی نیز همراه باشد. علائمی مانند عفونت مزمن گوش میانی، از دست دادن حس شنوایی، اختلالات دندانی یا بدفرمی دندانها، زخم شدن کبد (فیبروز کبدی)، آسم، ناهنجاریهای مادرزادی قلب، غیرطبیعی بودن موقعیت اندامهای داخلی و دیابت، کمتر در سندرم باردت بیدل رواج دارد.
ژنتیک مولکولی
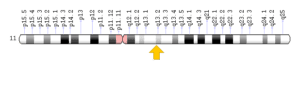
سندرم باردت بیدل در انسان توسط 12 ژن معیوب (BBS) در 11 کروموزوم غیرجنسی رخ میدهد که سه ژن جهشیافته در سه کروموزوم غیرجنسی، مشهودترین حالت این اختلال را نمایان میکنند. شایعترین ژن معیوب در سندرم باردت بیدل به دلیل جهش در ژن BBS1 است که در بازوی بلند کروموزوم شماره 11 بهصورت 11q13.2 مستقر است. این ژن در 30 درصد از موارد سندرم باردت بیدل انسانی دخالت دارد.
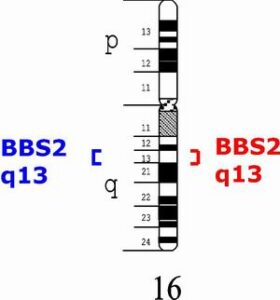
 دومین ژن معیوب شایع در سندرم باردت بیدل، ژن BBS2 است که در بازوی بلند کروموزوم شماره 16 بهصورت 16q21 مستقر است.
دومین ژن معیوب شایع در سندرم باردت بیدل، ژن BBS2 است که در بازوی بلند کروموزوم شماره 16 بهصورت 16q21 مستقر است.
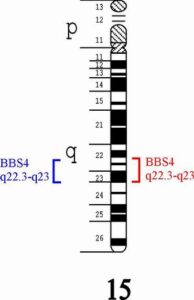
سومین ژن معیوب شایع در سندرم باردت بیدل، ژن BBS4 میباشد که در بازوی بلند کروموزوم شماره 15 بصورت 15q22.3-q23 مستقر است.
ژنهای شایع دیگر در سندرم باردت بیدل عبارتند از:
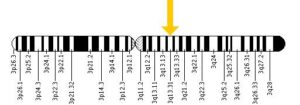
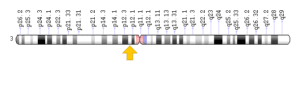
ژن BBS3 که در بازوهای بلند و کوتاه کروموزوم شماره 3 بصورت 3p12-q13 مستقر است.
ژن BBS5 که در بازوی بلند کروموزوم شماره 2 بصورت 2q31 مستقر است.

ژن BBS6 که در بازوی کوتاه کروموزوم شماره 20 بصورت 20p12 مستقر است.
ژن BBS7 که در بازوی بلند کروموزوم شماره 4 بصورت 4q27 واقع شده است.

ژن BBS8 که در بازوی بلند کروموزوم شماره 14 بصورت 14q32.12 واقع شده است.

ژن BBS9 که در بازوی کوتاه کروموزوم شماره 7 بصورت 7p14.3 واقع شده است.

ژن BBS10 که در بازوی بلند کروموزوم شماره 12 بصورت 12q21.2 مستقر است.

ژن BBS11 که در بازوی بلند کروموزوم شماره 9 بصورت 9q31.2-q34.1 واقع شده است.
ژن BBS12 که در روی بازوی بلند کروموزوم شماره 4 بصورت 4q27 واقع شده است.

محققان تعیین کردهاند که 20 تا 30 درصد از عوامل ژنتیکی سندرم باردت بیدل به دلیل جهش در یکی از این 12 ژن مذکور رخ میدهد، اما دلیل اصلی یا عامل اصلی که بیشترین استعداد ژنتیکی را در سندرم باردت بیدل موجب میشود هنوز ناشناخته است و احتمالاً ژنهای دیگری در این اختلال سهیم هستند.
پاتوفیزیولوژی
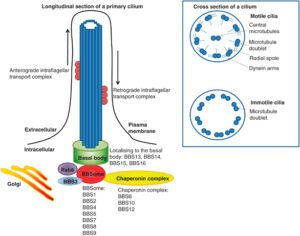
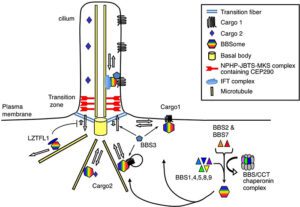
ژنهای مرتبط با سندرم باردت بیدل، هرکدام مسئول توسعه محصول پروتئینی خاصی هستند که جهش در هرکدام از آنها منجر به عدم سنتز پروتئین هدف یا سنتز اشتباه و نادرست از پروتئین موردنظر میشود. دانشمندان این محصولات پروتئینی را بهعنوان مژههای سلول معرفی میکنند، چرا که سلولها با این مژهها در ارتباط با یکدیگر هستند. محققان معتقدند که حرکت سلولها با این مژهها، منجر به تحرک مواد غذایی به داخل سلول میشود و این مژهها برای سلامت و رشد طبیعی سلول موردنیاز هستند.
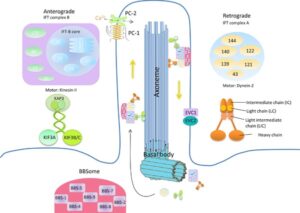
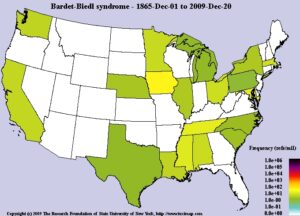
فرکانس
سندرم باردت بیدل مردان و زنان را به یک میزان تحتتأثیر قرار میدهد که بهصورت اتوزومال مغلوب به ارث میرسد. فرکانس این سندرم در مناطق مختلف جهان متفاوت است؛ بطوریکه در جمعیت شمال اروپا و آمریکا، فرکانس سندرم باردت بیدل 1 در 100000 میباشد، در سوئد فرکانس این اختلال 1 در 160000 تولد است، در جمعیت کویت 1 در 13500 تولد و در جمعیت نیوفاندلند 1 در 17500 تولد میباشد. این اختلال نخستین بار توسط دکتر باردت بیدل در سال 1920 گزارش گردید. سندرم لورنس موون که برخی از محققان معتقدند همان سندرم باردت بیدل است، اولین بار در سال 1865 توسط دکتر لورنس و موون گزارش گردید.
تشخیص
سندرم باردت بیدل معمولاً در دوران کودکی، بر اساس ارزیابی کامل بالینی و تشخیص یافتههای مشخصه مانند مشکلات بینایی ناشی از دیستروفی شبکیه، چاقی مفرط و پلیداکتیلی تشخیص داده میشود. بیماری شبکیه را میتوان با مراجعه به چشم پزشک و یا آزمایشهای اختصاصی از جمله ERG یا الکترورتینوگرام (روشی که با پالسهای الکتریکی، تحریک شبکیه به نور را اندازهگیری میکند) تشخیص داد. قطعیترین تست، آزمایش DNA است که اغلب ژنهای BBS1 و BBS10 را مورد بررسی قرار میدهند.

درمان
برای درمان سندرم باردت بیدل براساس نوع اختلال ایجاد شده، بیمار به متخصص قلب، متخصص دندان و فک و دهان، متخصص شنواییسنجی، متخصص بینایی، متخصص آسیبشناسی گفتار، متخصص اطفال، جراح ارتوپدی، و متخصص بیماریهای کلیه (نفرولوژیست) ارجاع داده میشود. در حال حاضر هیچ درمان قطعی برای دیستروفی شبکیه سندرم باردت بیدل وجود ندارد. در برخی موارد میتوان با جراحی دست و پا انگشتان اضافی را حذف کرد یا شکل معیوب آنها را تصحیح کرد.
با اعمال رژیمهای غذایی متناسب با سن بیماران و اعمال برنامههای ورزشی و تغییرات رفتاری میتوان میزان چاقی مفرط را در این مبتلایان مدیریت نمود. برای مردان هیپوگنادیسم مبتلا به سندرم باردت بیدل، میتوان با هورمون درمانی یا جایگزینی هورمون جنسی، میزان هیپوگنادیسم را کاهش داد. افراد مبتلا به سندرم باردت بیدل بایستی بطور مداوم تحت معاینه منظم متخصصان قرار گیرند تا در صورت تشخیص بهموقع برای اختلال کلیه، پیوند کلیه انجام گیرد و یا دیابت کنترل شود و همچنین عملکرد کبد و فشارخون بالا نیز مدیریت شود.
سندرم باردت بیدل، اولین بار توسط دو پزشک بنامهای جان ذکریا لورنس و رابرت چالرز موون در سال 1866 میلادی گزارش گردید که در آن زمان بنام بیماری لورنس موون شناخته میشد، اما با پیشرفت در زیستشناسی مولکولی و ژنتیک توسط دکتر باردت و بیدل به سندرم باردت بیدل تغییر نام داد.
:References
- Lewis RA. Bardet-Biedl Syndrome. NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia، PA. 2003:158-9.
- Gorlin RJ، Cohen MMJr، Hennekam RCM. Eds. Syndromes of the Head and Neck. 4th ed. Oxford University Press، New York، NY; 2001:1186-90.
- Jones KL. Ed. Smith’s Recognizable Patterns of Human Malformation. 5th ed. W. B. Saunders Co.، Philadelphia، PA; 1997:676-7.
- Tobin JL، Beales PL. Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007;22:926-36.
- Blacque OE، Leroux MR. Bardet-Biedl syndrome: an emerging pathomechanism of intracellular transport. Cell Mol Life Sci. 2006;63:2145-61.
- Moore SJ، Green JS، Fan Y، et al.، Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective، population-based، cohort study. Am J Med Genet A. 2005;132:352-60.
- Karmous-Benailly H، Martinovic J، Gubler MC، et al.، Antenatal presentation of Bardet-Biedl syndrome may mimic Meckel syndrome. Am J Hum Genet. 2005;76:493-504.
- Ansley SJ، Badano JL، Blacque OE، et al.، Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628-33.
- Barnett S، Reilly S، Carr L، et al.، Behavioral phenotype of Bardet-Biedl syndrome. J Med Genet. 2002;39:e76.
- Katsanis N، Ansley ST، Badano JL، et al.، Triallelic inheritance in Bardet-Biedl syndrome، a Mendelian recessive disorder. Science. 2001;293:2256-9.
- Katsanis N، Beales PL، Woods MO، et al.، Mutations in MKKS cause obesity، retinal dystrophy، and renal malformations associated with Bardet-Biedl syndrome. Nat Genet. 2000;26:67-70.
- Beales PL، Elcioglu N، Woolf AS، Parker D، Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437-46.
- Ross AJ، Beales PL. Updated:03/06/2007. Bardet-Biedl Syndrome. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright، University of Washington، Seattle. 1997-2003. Available at http://www.genetests.org.
- Beales P، Elcioglu N، Woolf A، Parker D، Flinter F (1 June 1999).
- Abd-El-Barr، MM; Sykoudis K; Andrabi S; Eichers ER; Pennesi ME; Tan PL; Wilson JH; Katsanis N; Lupski JR; Wu SM. (December 2007).
- Ansley SJ، Badano JL، Blacque OE، Hill J، Hoskins BE، Leitch CC، Kim JC، Ross AJ، Eichers ER، Teslovich TM، Mah AK، Johnsen RC، Cavender JC، Lewis RA، Leroux MR، Beales PL، Katsanis N (October 2003). “Basal body dysfunction is a likely cause of pleiotropic Bardet–Biedl syndrome”. Nature 425 (6958): 628–33.
- Blacque OE، Reardon MJ، Li C، McCarthy J، Mahjoub MR، Ansley SJ، Badano JL، Mah AK، Beales PL، Davidson WS، Johnsen RC، Audeh M، Plasterk RH، Baillie DL، Katsanis N، Quarmby LM، Wicks SR، Leroux MR (2004).
- Orozco، JT; Wedaman KP; Signor D; Brown H; Rose L; Scholey JM (1999). “Movement of motor and cargo along cilia”. Nature 398 (6729): 674.
https://medlineplus.gov/genetics/condition/bardet-biedl-syndrome/
برای دانلود پی دی اف بر روی لینک زیر کنید
ورود / ثبت نام
مطالب خیلی خوبی بود . لطفا من یه بیمار این مدلی دارم. ۸ ساله هست بینایی اش شب ها کم بینا هست . در مورد بینایی هیچ کاری نمیشه انجام داد؟
سلام از علایم دیگشون میگید لطفا