سندرم کمخونی دیاموند- بلکفان
Diamond-Blackfan Anemia Syndrome
شاهین اسعدی (کارشناس ارشد ژنتیک)

کلیاتی از سندرم کمخونی دیاموند- بلکفان
سندرم کمخونی دیاموند- بلکفان یک اختلال ژنتیکی است که در مغز استخوان تأثیر میگذارد. عملکرد اصلی مغز استخوان تولید سلولهای جدید است. در سندرم آنمی دیاموند- بلکفان، مغز استخوان دچار اختلال میشود و قادر به تولید گلبولهای قرمز کافی نیست تا اکسیژن را به بافتهای بدن انتقال دهند. کمبود گلبولهای قرمز معمولاً در طول سال اول زندگی ظاهر میشود. نشانههای کمخونی عبارتند از: خستگی، ضعف و رنگپریدگی پوست.
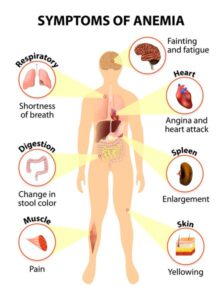
شکل 1: شماتیکی از علائم و نشانههای آنمی در ارگانهای بدن انسان
علائم و نشانههای بالینی سندرم کمخونی دیاموند- بلکفان
افراد مبتلا به سندرم کمخونی دیاموند- بلکفان در معرض خطر بیشتری از عوارض جدی مرتبط با مغز استخوان هستند. بهطور خاص افراد مبتلا به سندرم آنمی دیاموند- بلکفان در معرض خطر بیشتری نسبت به سندرم مایلودیسپلاستیک قرار دارند که طی آن رشد نرمال سلولهای نابالغ خون دچار اختلال میشود، علاوه بر این، افرادی که تحت تأثیر سندرم آنمی دیاموند- بلکفان قرار دارند دارای خطر افزایش ابتلا به برخی از سرطانها منجمله سرطان خون به نام لوسمی حاد مایلوئیدی (AML) و یک نوع سرطان استخوان به نام اُستئوسارکوم هستند.

شکل 2: شماتیکی از سلولهای خون نرمال در مقابل سلولهای مبتلا به آنمی
تقریباً نیمی از افراد مبتلا به سندرم آنمی دیاموند- بلکفان اختلالات فیزیکی دارند. آنها ممکن است دارای اندازه سر بسیار کوچک (میکروسفالی)، خط موی کمپشت در پوست سر، فاصله گسترده چشمها از یکدیگر (هایپرتلوریسم)، افتادگی پلکی (پتوزیس)، پُل پهن و صاف بینی، گوشهای کوچک و فک پایین کوچکتر از حد نرمال (میکروگناتیا) باشند، همچنین افراد آسیبدیده ممکن است دارای شکاف کام و شکاف لب باشند. آنها ممکن است گردن کوتاه داشته باشند و تیغه شانه نیز کوچکتر و بالاتر از حد نرمال است. شایعترین نشانه سندرم آنمی دیاموند- بلکفان ناهنجاری و بدشکلی دستها در مبتلایان است. حدود یکسوم افراد مبتلا به سندرم آنمی دیاموند- بلکفان رشد آهستهای دارند که منجر به کوتاهی قد آنها خواهد شد.

شکل 3: تصویر کودک مبتلا به سندرم آنمی دیاموند- بلکفان همراه با ویژگیهای متمایز صورت
سایر ویژگیهای سندرم آنمی دیاموند- بلکفان ممکن است شامل مشکلات چشم مانند کِدِر شدن چشمها (آبمروارید)، افزایش فشار داخلی چشم (گلوکوم) و یا لوچی چشم (استرابیسم) باشد. افراد آسیبدیده ممکن است دارای اختلالات کلیوی و همچنین نقص ساختاری قلب نیز باشند و در مردان قرار گرفتن مجاری اِدراری در زیر آلت تناسلی (هایپوسپادیاس) نیز ممکن است رخ دهد.
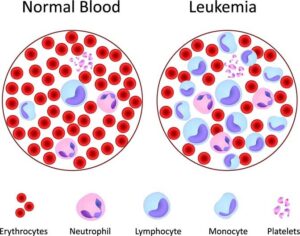
شکل 4: نمای شماتیک از سلولهای خون نرمال در مقابل سلولهای مبتلا به لوسمی
علتشناسی سندرم کمخونی دیاموند- بلکفان
سندرم آنمی دیاموند- بلکفان میتواند در اثر جهش ژن RPL5 که در بازوی کوتاه کروموزوم شماره 1 بهصورت 1p22.1 مستقر است، ژن RPL11 که در بازوی کوتاه کروموزوم شماره 1 بهصورت 1p36.11 مستقر است، ژن RPL35A که در بازوی بلند کروموزوم شماره 3 بهصورت 3q29 مستقر است، ژن RPS7 که در بازوی کوتاه کروموزوم شماره 2 بهصورت 2p25.3 مستقر است، ژن RPS10 که در بازوی کوتاه کروموزوم شماره 6 بهصورت 6p21.31 مستقر است، ژن RPS17 که در بازوی بلند کروموزوم شماره 15 بهصورت 15q25.2 مستقر است، ژن RPS19 که در بازوی بلند کروموزوم شماره 19 بهصورت 19q13.2 مستقر است، ژن RPS24 که در بازوی بلند کروموزوم شماره 10 بهصورت 10q22.3 مستقر است و ژن RPS26 که در بازوی بلند کروموزوم شماره 12 بهصورت 12q13.2 مستقر است، ایجاد شود. این ژنها دستورالعملهای لازم برای سنتز چندین پروتئین از ریبوزوم 80s را فراهم میکنند. ریبوزومها دستورالعملهای ژنتیکی سلول را برای تولید پروتئین پردازش میکنند.
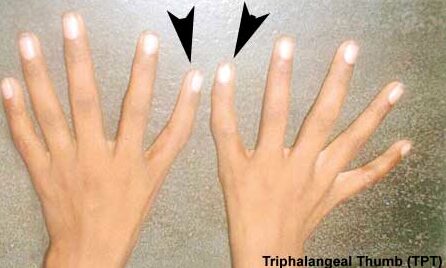
شکل 5: تصویر دستهای فرد مبتلا به سندرم آنمی دیاموند- بلکفان همراه با ناهنجاری انگشتان
هر ریبوزوم از دو زیرواحد بزرگ و کوچک ساخته شده است. ژنهای RPL5 ,RPL11 ,RPL35A دستورالعملهای لازم برای سنتز پروتئینهای ریبوزومی زیرواحد بزرگ را فراهم میکنند. ژنهـــــای RPS7 ,RPS10 ,RPS17 ,RPS19 ,RPS24 ,RPS26، دستورالعملهای لازم برای سنتز پروتئینهای ریبوزومی زیرواحد کوچک را فراهم میکنند.
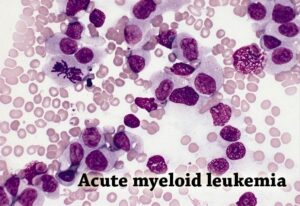
شکل 6: تصویر میکروسکوپیک از گستره خون مبتلا به لوسمی حاد مایلوئیدی
عملکرد دقیق پروتئینهای ریبوزومی در این زیرواحد بزرگ و کوچک مشخص نیست. برخی از پروتئینهای ریبوزومی در مونتاژ یا پایداری ریبوزومها دخیل هستند. سایر پروتئینهای ریبوزومی کمک میکنند تا ریبوزومها، پروتئینهای جدید را بهطور صحیح مونتاژ کنند. مطالعات نشان میدهد که برخی از پروتئینهای ریبوزومی ممکن است در توابع دیگری مانند شرکت در مسیرهای سیگنالینگ بیوشیمیایی در داخل سلول، تنظیم سلولها و کنترل سلولهای آپوپتوزی نقش داشته باشند.
اعتقاد بر این است که جهش در هر یک از ژنهای ذکرشده در بالا به ثبات یا عملکرد پروتئینهای ریبوزومی تأثیر میگذارد. مطالعات نشان میدهد که کمبود پروتئینهای ریبوزومی عملکردی ممکن است باعث از بین بردن سلولهای تشکیلدهنده خون در مغز استخوان شده و منجر به کمخونی شود. تنظیم غيرطبيعی تقسيم سلولی يا تحريک نامناسب آپوپتوز ممكن است به مشكلات سلامتی دیگری كه در بعضی افراد مبتلا به کمخونی دیاموند- بلکفان تأثیر میگذارد، منجر شود.
تقریباً 25 درصد از افراد مبتلا به کمخونی دیاموند- بلکفان جهش در ژن RPS19 را دارند. حدود 25 تا 35 درصد افراد مبتلا به این اختلال جهش در ژنهای RPL5، RPL11، RPL35A، RPS7، RPS10، RPS17، RPS24 یا RPS26 را دارند. در 40 تا 50 درصد باقیمانده، علت بیماری ناشناخته است. محققان معتقدند که ژنهای دیگر نیز ممکن است با کمخونی دیاموند- بلکفان ارتباط داشته باشند.

شکل 7: نمای شماتیک از کروموزوم شماره 1 که ژن RPL5 در بازوی کوتاه این کروموزوم بهصورت 1p22.1 مستقر است
 شکل 8: نمای شماتیک از کروموزوم شماره 1 که ژن RPL11 در بازوی کوتاه این کروموزوم بهصورت 1p36.11 مستقر است
شکل 8: نمای شماتیک از کروموزوم شماره 1 که ژن RPL11 در بازوی کوتاه این کروموزوم بهصورت 1p36.11 مستقر است

شکل 9: نمای شماتیک از کروموزوم شماره 3 که ژن RPL35A در بازوی بلند این کروموزوم بهصورت 3q29 مستقر است

شکل 10: نمای شماتیک از کروموزوم شماره 2 که ژن RPS7 در بازوی کوتاه این کروموزوم بهصورت 2p25.3 مستقر است

شکل 11: نمای شماتیک از کروموزوم شماره 6 که ژن RPS10 در بازوی کوتاه این کروموزوم بهصورت 6p21.31 مستقر است

شکل 12: نمای شماتیک از کروموزوم شماره 15 که ژن RPS17 در بازوی بلند این کروموزوم بهصورت 15q25.2 مستقر است
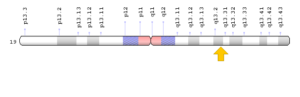
شکل 13: نمای شماتیک از کروموزوم شماره 19 که ژن RPS19 در بازوی بلند این کروموزوم بهصورت 19q13.2 مستقر است

شکل 14: نمای شماتیک از کروموزوم شماره 10 که ژن RPS24 در بازوی بلند این کروموزوم بهصورت 10q22.3 مستقر است

شکل 15: نمای شماتیک از کروموزوم شماره 12 که ژن RPS26 در بازوی بلند این کروموزوم بهصورت 12q13.2 مستقر است
شکل 16: شماتیکی از ساختار اندامک ریبوزوم
سندرم آنمی دیاموند- بلکفان از الگوی توارثی اتوزومال غالب پیروی میکند، بنابراین برای ایجاد این سندرم، یک نسخه از ژنهای جهشیافته ذکرشده (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به این سندرم در حالت اتوزومی غالب برای هر بارداری احتمالی به میزان 50% است.
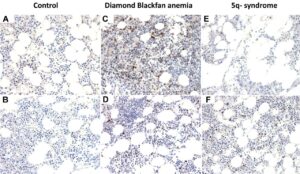
شکل 17: تصویر میکروسکوپیک از اختلالات آنمی دیاموند- بلکفان (سمت وسط)، سندرم حذفی بازوی بلند کروموزوم شماره 5 (سمت راست) در مقابل گستره خون سالم (سمت چپ)
فراوانی سندرم کمخونی دیاموند- بلکفان
سندرم کمخونی دیاموند- بلکفان اختلال ژنتیکی بسیار نادری است که فرکانس شیوع آن در جهان حدود 5 تا 7 در هر 1 میلیون تولد زنده از سراسر جهان است.

شکل 18: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم آنمی دیاموند- بلکفان از این الگو تبعیت میکند
تشخیص سندرم کمخونی دیاموند- بلکفان
سندرم کمخونی دیاموند- بلکفان بر اساس یافتههای بالینی و کلینیکی مبتلایان و برخی آزمایشهای پاتولوژیکی، تشخیص داده میشود. دقیقترین روش تشخیص این سندرم، آزمایش ژنتیک مولکولی برای ژنهای مذکور بهمنظور بررسی وجود جهشهای احتمالی است.
مسیرهای درمانی سندرم کمخونی دیاموند- بلکفان
استراتژی درمان و مدیریت سندرم کمخونی دیاموند- بلکفان بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص هماتولوژی، متخصص ارتوپدی، متخصص چشم، متخصص پوست و مو و زیبایی، متخصص گوش و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان استانداردی برای این سندرم وجود ندارد و تمامی اقدامات بالینی بهمنظور تخفیف رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند، از اهمیت بسزایی برخوردار است.
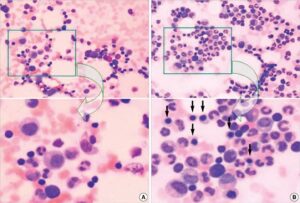
شکل 19: تصویر میکروسکوپیک از گستره خونی در فرد مبتلا به سندرم آنمی دیاموند- بلکفان
تاریخچه سندرم کمخونی دیاموند- بلکفان
این اختلال اولین بار در سال 1936 توسط Hugh W.Josephs توصیف شد، سپس در سال 1938 توسط دکتر Louis K.Diamond و Kenneth Blackfan بهطور کاملتر گزارش گردید.
جدول شماره 1: انواع سندرم کمخونی دیاموند- بلکفان همراه با ژنها و پروتئینهای مربوطه با جایگاه کروموزومی و اختلالات مربوط به سایتهای ریبوزومی
| نام | جایگاه کروموزومی | شماره ژنوتیپ در OMIM | شماره فنوتیپ در OMIM | پروتئین | اختلال |
| DBA1 | 19q13.2 | 603474 | 105650 | RPS19 | 30S to 18S |
| DBA2 | 8p23-p22 | unknown | 606129 | ||
| DBA3 | 10q22-q23 | 602412 | 610629 | RPS24 | 30S to 18S |
| DBA4 | 15q | 180472 | 612527 | RPS17 | 30S to 18S |
| DBA5 | 3q29-qter | 180468 | 612528 | RPL35A | 32S to 5.8S/28S |
| DBA6 | 1p22.1 | 603634 | 612561 | RPL5 | 32S to 5.8S/28S |
| DBA7 | 1p36.1-p35 | 604175 | 612562 | RPL11 | 32S to 5.8S/28S |
| DBA8 | 2p25 | 603658 | 612563 | RPS7 | 30S to 18S |
| DBA9 | 6p | 603632 | 613308 | RPS10 | 30S to 18S |
| DBA10 | 12q | 603701 | 613309 | RPS26 | 30S to 18S |
| DBA11 | 17p13 | 603704 | 614900 | RPL26 | 30S to 18S |
| DBA12 | 3p24 | 604174 | 615550 | RPL15 | 45S to 32S |
| DBA13 | 14q | 603633 | 615909 | RPS29 | |
| سایر |
SLC49A1 (FLVCR1) |
منبع:
اسعدی شاهین، دکتر قلیزاده شیوا، دکتر روشن روان ندا، دکتر علیپور شهریار، محمدزاده حمیده، ولیزاده گلنساء، اَمجدی حسین، جمالی مهسا، کتاب پاتولوژی در ژنتیک پزشکی جلد 9 (A-H)، فصل چهارم (D)، صفحات 297-285، انتشارات کتب دانشگاهی عمیدی، پائیز 1397.
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام