سندرم اُولیئر
Ollier Syndrome
شاهین اسعدی (کارشناس ارشد ژنتیک)، ندا غفوری (کارشناس ارشد بیوشیمی)، مهسا جمالی (کارشناس ارشد ژنتیک)

نگارنده مسئول: شاهین اسعدی
کلیات
سندرم اُولیئر یک اختلال ژنتیکی است که توسط اِنکوندرومهای چندگانه مشخص میشود که بصورت رشد غده سرطانی غضروف در داخل استخوانها است. این رشد اغلب در استخوانهای اندامها بهویژه در استخوانهای دست و پا رخ میدهد، با این حال ممکن است در جمجمه، دندهها و استخوانهای ستون فقرات (مهرهها) نیز رخ دهد. اِنکوندرومها ممکن است منجر به ناهنجاریهای شدید استخوانی، کوتاه شدن اندامها و پوسیدگی شوند.
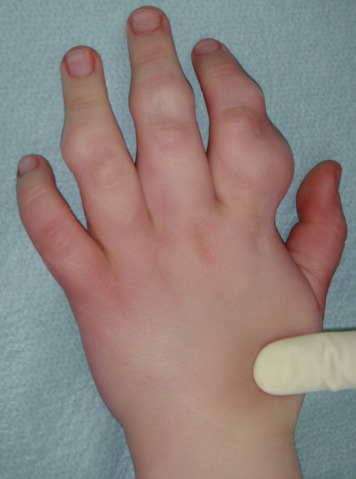
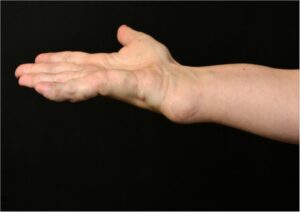
شکل 1: تصویر دست مبتلا به سندرم اُولیئر همراه با اختلال بافت استخوانی اِنکوندروم
علائم و نشانههای بالینی
علائم و نشانههای سندرم اُولیئر ممکن است در هنگام تولد تشخیص داده شود، اگرچه معمولاً تا سن 5 سالگی آشکار نمیشوند. اِنکوندرومها در نزدیکی انتهای استخوان که در آن رشد طبیعی رخ میدهد، ایجاد میشوند و اغلب پس از شروع، رشد افراد در اوایل بزرگسالی متوقف میشود، در نتیجه افراد مبتلا به این سندرم معمولاً دارای رشد کم و کوتاهی قد هستند.
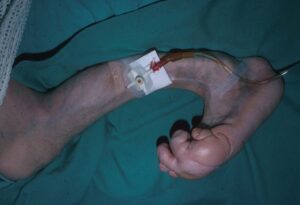
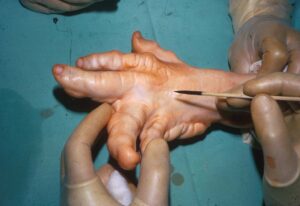
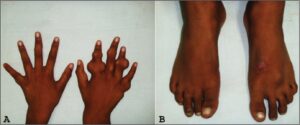
شکل 2: نمایی دیگر از دستها و پاهای انسان مبتلا به سندرم اُولیئر همراه با اختلالات بافت استخوانی
اگرچه اِنکوندرومهای مرتبط با سندرم اُولیئر بهصورت خوشخیم هستند، اما ممکن است تبدیل به سرطان (بدخیم) شوند. بهطور خاص، افراد مبتلا ممکن است به سرطانهای استخوانی به نام کوندروسارکوم را بهویژه در جمجمه دچار شوند. شایان ذکر است که ابتلاء به این سندرم خطر ابتلا به سرطانهای دیگر مانند سرطان تخمدان و کبد را افزایش میدهد.
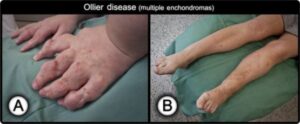
شکل 3: تصاویری از اختلالات بافت استخوانی اِنکوندروم در دستها و پاهای مبتلا به سندرم اُولیئر
افراد مبتلا به سندرم اُولیئر معمولاً عمر نرمال دارند و بهره هوشی تحت تأثیر این سندرم قرار نمیگیرد. میزان آسیب فیزیکی آنها بستگی به انحطاط اسکلتی فرد دارد، اما در اغلب موارد آنها در فعالیتهای حرکتی خود محدودیتهای عمدهای ندارند.
علت شناسی
اغلب موارد سندرم اُولیئر در اثر جهش ژن IDH1 که در بازوی بلند کروموزوم شماره 2 بهصورت 2q34 مستقر است و یا ژن IDH2 که در بازوی بلند کروموزوم شماره 15 بهصورت 15q26.1 مستقر است، ایجاد میشود. این ژنها دستورالعملهای لازم برای سنتز آنزیمهایی به نام ایزوسیترات دهیدروژناز 1 و ایزوسیترات دهیدروژناز 2 را فراهم میکنند. این آنزیمها ترکیبی به نام ایزوسیترات را به یک ترکیب دیگر تحت عنوان 2-کتوگلوتارات تبدیل میکنند. این واکنش نیز مولکولی به نام NADPH تولید میکند که برای بسیاری از فرآیندهای سلولی ضروری است.
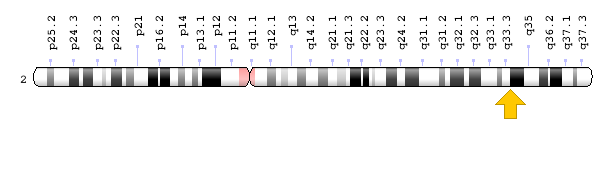
شکل 4: نمای شماتیک از کروموزوم شماره 2 که ژن IDH1 در بازوی بلند این کروموزوم بهصورت 2q34 مستقر است
جهشهای ژن IDH1 یا IDH2 باعث تولید آنزیم غیرطبیعی و یا عدم تولید آنزیم ایزوسیترات دهیدروژناز 1 میشوند. هنوز ارتباط بین این جهشها و علائم این سندرم بهخوبی درک نشده است.
سندرم اُولیئر از هیچ الگوی توارثی تبعیت نمیکند و این سندرم در اثر جهشهای جدید سوماتیکی ایجاد میشود. شایان ذکر است که در این سندرم ، جهش در برخی از سلولها وجود دارد و در برخی وجود ندارد و به این حالت، موزائیسم گفته میشود.
فراوانی
سندرم اُولیئر اختلال ژنتیکی است که فرکانس شیوع آن در جهان حدود 1 در 100000 نفر برآورد شده است.

شکل 5: نمای شماتیک از کروموزوم شماره 15 که ژن IDH2 در بازوی بلند این کروموزوم بهصورت 15q26.1 مستقر است

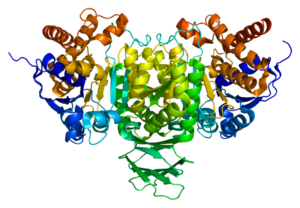
شکل 6: شماتیکی از توالی اگزونی و ساختمانی پروتئینهای IDH1 و IDH2
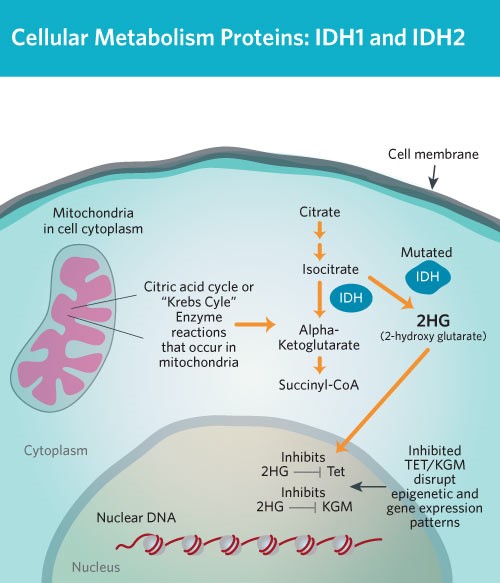
شکل 7: شماتیکی از متابولیسم سلولی پروتئینهای IDH1 و IDH2
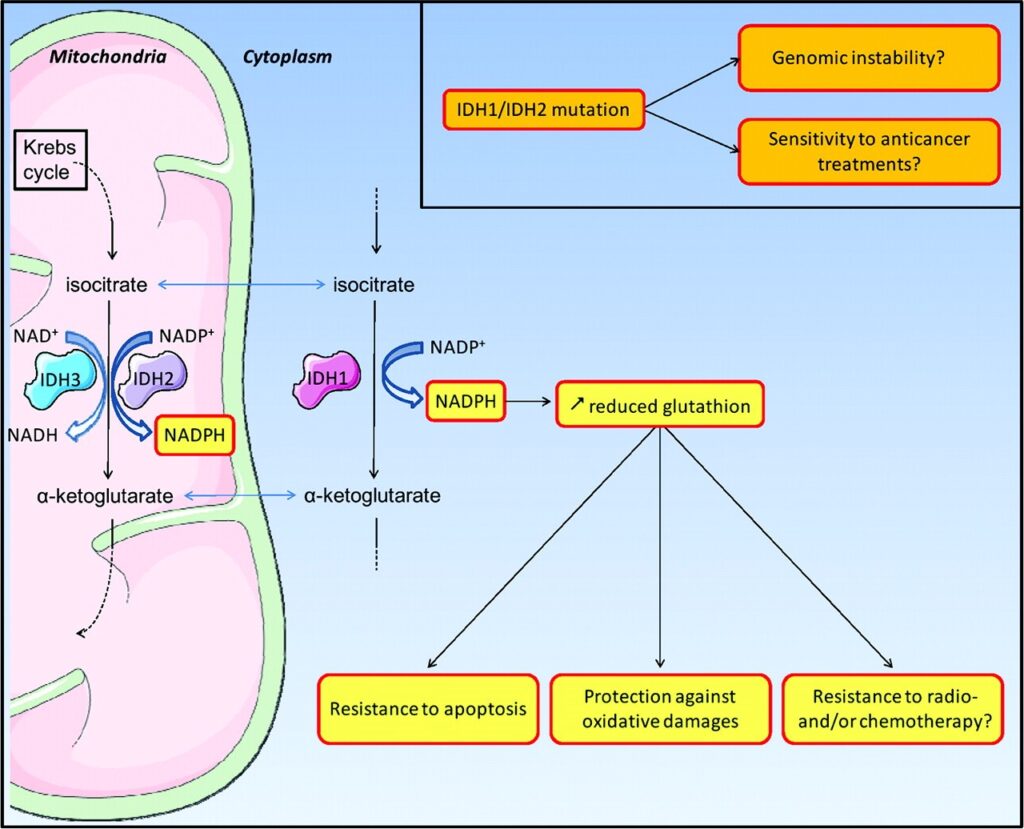
شکل 8: شماتیکی از عملکرد ژنهای جهشیافته IDH1 و IDH2
تشخیص
سندرم اُولیئر بر اساس یافتههای بالینی، فیزیکی و کلینیکی مبتلایان و برخی آزمایشهای پاتولوژیکی تشخیص داده میشود. دقیقترین روش تشخیص این سندرم، آزمایش ژنتیک مولکولی برای ژنهای IDH1 و IDH2 بهمنظور بررسی وجود جهشهای احتمالی میباشد. تشخیص پیش از تولد نیز با استفاده از تکنیک PGD و مایع آمنیوسنتز و یا نمونهبرداری از پرزهای کوریونی جفت جنین امکانپذیر است.
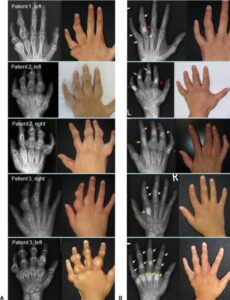
شکل 9: تصاویری از اختلالات بافت استخوانی اِنکوندروم در دستهای مبتلا به سندرم اُولیئر
مسیرهای درمانی
استراتژی درمان و مدیریت این سندرم بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص ارتوپدی، متخصص تغذیه، جراحان و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان قاطعی برای این سندرم وجود ندارد و تمامی اقدامات بالینی بهمنظور تخفیف رنج مبتلایان میباشد. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند از جایگاه ویژهای برخوردار است.
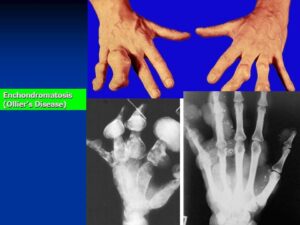
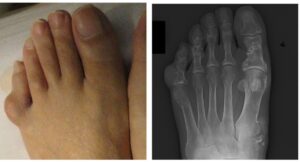
شکل 10: نمای رادیولوژیکی از دستها و پاهای مبتلا به سندرم اُولیئر
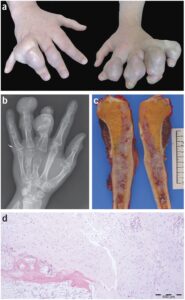
شکل 11: تصاویر رادیولوژیکی و میکروسکوپیک از بافت استخوانی اِنکوندروم در سندرم اُولیئر
منبع:
اسعدی شاهین، قلیزاده شیوا، روشن روان ندا، علیپور شهریار، محمدزاده حمیده، ولیزاده گلنساء، امجدی حسین، جمالی مهسا، کتاب پاتولوژی در ژنتیک پزشکی 10 (I-X)، فصل 15، صفحات 561-551، انتشارات کتب عمیدی، 1397.
مورفولوژی و عملکرد سلولهای استخوانی
https://rarediseases.org/rare-diseases/ollier-disease/
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام