انواع مختلف PCR
دکتر میرنژاد
در کارهای تشخیصی در آزمایشگاههای بالینی از روشهای مختلف PCR استفاده میگردد که این روش عبارتند از:
- Touchdown PCR
- Degenearte PCR
- Heminested PCR
- RT–PCR
- ARMS-PCR
- Nested-PCR
- Hot-Start PCR
- Alu-PCR
- PCR-ELISA
- Real –Time PCR
- Multiplex PCR
- و ….
در ادامه به بعضی از این نوع PCRها اشاره کوتاه میگردد.:
Multiplex-PCR
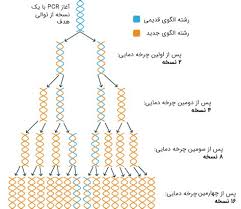
Multiplex PCR یک نوع تغییر یافته از PCR میباشد که در آن دو یا بیش از دو لوکوس از ژن به طور همزمان در یک واکنش آمپلی میشود. از سال 1988 که برای اولین بار این روش توصیف شد بطور موفقیتآمیزی در خیلی از تستها شامل آنالیز جهش ناشی از حذف، سایر جهشها، پلیمورفیسم یا سنجش کمی بکار رفته است. امروزه از این روش در شناسایی پاتوژنها، اسکرین جنسیت، آنالیز Linkage، مطالعات جرمشناسی، بررسی کیفیت و کمیت نمونهها و تشخیص بیماریهای ژنتیکی استفاده میگردد.
بطورکلی در شناسائی پاتوژنها، Multiplex PCR باکتریها نشان دهنده یک پاتوژن خاص در میان دیگر باکتریها یا تمایز گونهها یا سویهها در یک جنس می باشد. در Multiplex PCR صرف زمان و معرف نسبت به PCR معمولی که در چندین لوله انجام میشود کمتر میباشد به همین دلیل واکنش مولتیپلکس برای استفاده کم از آنزیم و الگوها بسیار مناسب است.
طراحی و اجرای Multiplex PCR
در Multiplex PCR میتوان دو مجموعه از پرایمرها که شرایط واکنش آنها بطور جداگانه ارزیابی شده است را با هم مخلوط کرد. هرچند در بعضی Multiplex PCR با توجه به ملاحظات دقیق برای نواحی که آمپلی میشوند، اندازه نسبی قطعات، دینامیک پرایمرها و اپتمایز کردن تکنیک PCR با چندین قطعه مناسب تغییراتی داده شده است. مانند تمام انواع PCR باید در Multiplex PCR اهداف را مشخص کرد و با توجه به آنها پرایمرها را طراحی نمود (شکل 1). این پرایمرها بایستی دارای کنتیک واکنش مشابهای باشند، مقدار C/G پرایمرها حدود 60-40%، طول آنها 28-23 نوکلئوتید و درجه حرارت annealing متوسط داشته باشند.
درجه حرارت annealing اولیه و غلظت مواد باید ابتدا ارزیابی شوند، هرچند که این شرایط را میتوان در Multiplex PCR بصورت تجربی بدست آورد. به همین دلیل شرایط برای هر مجموعه پرایمر میبایستی بطور جداگانه طراحی شود و اگر در مجموعه های پرایمر نیاز به اصلاح بود تغییرات اعمال گردد .
جفتهای پرایمر در هنگامی که جدا کار میکنند مشکلی ندارند، ولی وقتی به صورت ترکیبی بکار میروند مشکل بوجود میآید که برای رفع مشکلات میتوان از اتانول مناسب در استات سدیم 0/3 مولار استفاده کرد. احتمال تشکیل جفتهای غیراختصاصی و آرتیفکتها در اضافه کردن هر مجموعه پرایمر افزایش پیدا میکند. برای رفع باندهای نامناسب Multiplex PCR میتوان از Hot-start PCR، اضافه کردن مواد آلی، بالا بردن درجه حرارت Annealing استفاده کرد و همچنین اگر از موارد فوق استفاده شد و باندهای نامناسب هنوز وجود داشت بایستی پرایمر جدید طراحی شود.
اگر در غلظتهای اکیمولار محصول بدست نیامد و برای همه قطعات آمپلیفیکاسیون صورت نگرفت، بایستی غلظت بعضی از جفت پرایمرها را کاهش داد. این تغییر به خصوص در نمونههائی که یک هدف بیش از دیگر اهداف دارند مفید میباشد. ارتباط معکوسی بین غلظت الیگونوکلئوتید مورد نیاز در Multiplex PCR و مقدار ATP آنها وجود دارد، اما ارتباطی بین موارد فوق با طول الیگونوکلئوتید و Tm وجود ندارد.
هنگامی که بین همه جفت پرایمرها سازگاری وجود ندارد، بایستی آنها را در چندین واکنش Multiplex PCR کوچکتر بررسی کرد. مثلاً در یک مطالعه در ابتدا 18 جفت پرایمر و سپس با اپتیمایز کردن شرایط واکنش با 26 جفت پرایمر برای شناسائی ژن دیستروفی در Multiplex PCR استفاده شد.

شکل (1): نمای شماتیک از Multiplex PCR
تیتراسیون اجزای واکنش:
در PCR به خصوص در Multiplex PCR ضروری است که غلضتهای مختلف اجزای واکنش برای بدست آوردن بالاترین کارآئی روش PCR با همدیگر متناسب شوند. غلظت Mg2+ و dNTP و آنزیم پلیمراز اغلب روی تعداد آمپلیکون در مالتیپلکس اثر میگذارند، لذا مانند روش PCR معمولی، غلظتها بایستی اپتیمایز شوند، چراکه جفت پرایمرها ممکن است دارای نیازمندیهای متفاوت باشند. سیستمهای بافری بطور خیره کننده در آمپلیفیکاسیون موثر هستند. از DMSO میتوان به عنوان بازدارنده یا اجزای مفید در Multiplex PCR استفاده کرد. مواد افزودنی که سبب کاهش باندهای غیر اختصاصی در Multiplex PCR میشوند عبارتنداز: توئین20، تریتون 100-X، ß مرکاپتواتانول و کلرید تترامتیل آمونیم.
شرایط ترموسایکلر:
پارامترهای ترموسایکلر با سکانس مجموعههای پرایمر تعیین میشود. عموماً هرچه تعداد اهداف بیشتر باشد زمان بیشتری باید برای انجام PCRدر نظر گرفته شود. هرچند که در اینجا بایستی این نکته را در نظر داشت که هرچه زمانAnnealing و Extension طولانیتر شود فرصت برای آمپلیفیکاسیون غیراختصاصی هم افزایش مییابد.
PCR-ELISA
میتوان این روش را جایگزین Real –Time PCR استفاده کرد. محصولات PCR بوسیله مواد نشاندار )مانند digoxigenin) در طی آمپلی کردن نشاندار میشود. سپس این محصول نشاندار با یک پروپ اختصاصی محصول PCR که در دیواره میکروپلیت متصل است باند میشود. در مرحله بعد آنتیبادی متصل به آنزیم ضد digoxigenin برای ارزیابی مقادیر محصول PCR مورد استفاده قرار میگیرد. میتوان بجای digoxigenin از بیوتین استفاده کرد. این روش در مقایسه با PCR معمولی حساستر، اختصاصیتر، ارزانتر و سریعتر میباشد(شکل2).

شکل (2): سنجش PCR-ELISA
Nested-PCR
روش موفقي جهت از بين بردن محصولات ناخواسته و افزايش همزمان حساسيت PCR ميباشد. اساس اين روش ابتدا تکثیر قطعهاي DNA مورد نظر تحت شرايط استاندارد با دو پرایمر اولیه میباشد که در مرحله بعد محصول PCR اولیه با استفاده از پرايمرهاي داخليتر نسبت به جايگاههاي دو پرايمر اوليه PCR میگردد (شکل 3). در این روش ابتدا پرایمر بیرونی توالی هدف در طول 30-15چرخه تکثیر میشود، سپس محصول PCR حاصل به لولهای دیگر منتقل میشود و به عنوان الگو و با استفاده از جفت پرایمر داخلی مرحله دوم PCR انجام شده و ترادف کوچکتری از DNA که درون PCR اولی است، به اندازه 40-15 چرخه تکثیر میگردد.
در این روش به دلیل انتقال محصول PCR دور اول به لوله جدید، بازدارندهها رقیق میشوند. اختصاصیت PCR را ویژگی پرایمرها تعیین میکنند. برای مثال اگر پرایمرها در یک مخلوط بزرگ DNA ژنومی به بیشتر از یک لوکوس متصل شوند، بیشتر از یک قطعه DNA تکثیر خواهد شد. به همین جهت محققان سعی میکنند که از Nested Primers بخاطر تأمین اختصاصیت استفاده کنند.

شکل (3): نمای شماتیک از Nested-PCR
Alu-PCR
در اين روش كه بيشتر براي تعيين تواليهاي كروموزومي انسان در سلولهاي هيبريدي استفاده ميشود از پرايمرهايي كه مكمل تواليهاي بسيار تكراري ژنومها ميباشد (اين تواليهاي 300 جفت بازي كه گاهي تا نهصد هزار بار در ژنوم انسان و ديگر پستانداران تكرار ميشوند را تكرارهاي ALu مينامند) استفاده ميگردد.
روشهای عملی در Time PCR – Real قسمت5
https://en.wikipedia.org/wiki/Variants_of_PCR
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام