
آزمایشگاه و بالین
نقش NGS در تحقیقات سرطان
و کاربرد بالینی آن
(قسمت اول)

دکتر محسن منشدی
آزمایشگاه تشخیص طبی دکتر منشدی
چکیده
کاربرد گستردهی NGS، بهطور عمده از میان غربالگری کل ژنوم، اگزوم و ترانسکریپتوم، اطلاعات باارزشی را از وضیعت سلولهای سرطانی در اختیار ما قرار میدهد. همراه با استفاده از ابزار بیوانفورماتیک، NGS انقلابی در تحقیق، تشخیص و درمان سرطان ایجاد کرده است. در این مقاله، مروری بر پیشرفتهای اخیر در زمینه تحقیقات ژنومی سرطان مبتنی بر NGS و همچنین کاربردهای بالینی آن داریم و بهطور خلاصه با توضیح پروژههای انکوژنومیک، منابع و الگوریتمهای مربوطه، به بحث در مورد مشکلات مرتبط با تحقیقات در این حوزه میپردازیم.
مقدمه
طی دو دههی اخیر، “تعیین توالی سانگر”در تحقیقات ژنومی بیشترین نقش را ایفا کرده و دستاوردهای زیادی داشته است، از جمله تعیین توالی ژنوم انسانی که منجر به شناسایی اختلالات منوژنیک شد و همچنین بررسی جهشهای هدفمند ایجادشده در سلولهای سوماتیک را امکانپذیر کرد (1 و 2). علیرغم دستاوردهای باارزش تعیین توالی سانگر، نیاز به روشهای تعیین توالی با سرعت بالا و قیمت پایین، از دغدغههای اساسی محققین بود که منجر به ظهور روشی به نام NGS[1] گردید. توانایی NGS برای فراهم ساختن حجم بالایی از اطلاعات با قیمت پایین (3 و 4) این امکان را برای محققین فراهم میکند تا چشمانداز مولکولی انواع مختلف سرطان را تعیین کنند و به پیشرفتهای چشمگیری در مطالعات ژنومی سرطان دست یابند.
استفاده از NGS در قالب بررسی کل ژنوم (WGS) و یا کل اگزونها (WES)، باعث ایجاد انفجاری در اطلاعات مربوط به تغییرات سلولهای سرطانی همچون موتاسیونهای نقطهای، درج یا حذفهای کوچک، تنوع در تعداد کپیهای تکراری و تغییرات ساختاری شده است. با مقایسهی این تغییرات با نمونههای طبیعی، محققین قادر به تشخیص این تغییرات در رده سلولهای جنسی و سوماتیک شدهاند. با استفاده از این روش برای تعیین توالی ترانسکریپتوم، علاوه بر کسب اطلاعات باارزشی در خصوص میزان بیان ژن، میتوان از ایزوفرمهای اسپلیسینگ mRNA موجود در سلول، آگاهی پیدا کرد. همچنین، تغییرات اپیژنتیکی، تغییر متیلاسیون DNA و تغییرات هیستون با استفاده از روشهای دیگر تعیین توالی، مانند توالی بیسولفیت و ChIP- seq.، مورد مطالعه قرار میگیرند. استفاده از روشهای فوقالذکر اطلاعات باارزشی را در مورد ژنوم سلول سرطانی در اختیار ما قرار میدهد. با استفاده از ابزارهای قدرتمند بیوانفورماتیک، امکان درک بهتر بیولوژی سرطان و توسعهی روشهای درمانی طراحیشده، وجود دارد.شکل 1 چگونگی ارتباط روشهای مختلف در تحقیقات سرطان و استفادهی بالینی از آنها را نشان میدهد.
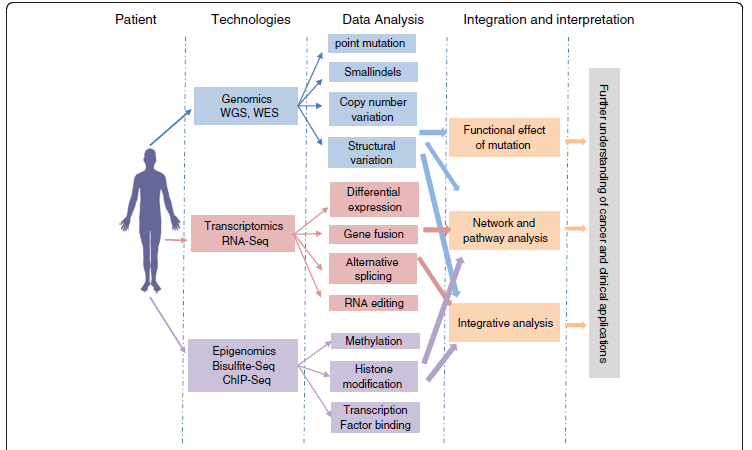
شکل 1: گردش کار حاصل از اطلاعات omics در تحقیقات سرطان و کاربرد بالینی
فنآوریهای NGS قادر است تغییرات ژنومیک، ترانسکریپتومیک و اپیژنومیک مشتمل بر جهشها، تنوع تعداد کپی، انواع تغییرات ساختاری و بیان ژن، همجوشی ترانسکریپتها، تغییر متیلاسیون DNA و غیره را شناسایی کند. در این رابطه، انواع مختلف ابزارهای بیوانفورماتیک برای تجزیه و تحلیل و تفسیر دادهها بهمنظور درک بهتری از بیولوژی سرطان و توسعه استراتژی درمانهای اختصاصی بکار میروند.
تحقیقات سرطان
در چند سال گذشته، بسیاری از مطالعات بر پایه NGS بهمنظور توصیف جامع و دقیق تغییرات مولکولی سرطانها، شناسایی تغییرات مؤثر در سرطانزایی، پیشرفت سرطان، ایجاد متاستازها و همچنین بهمنظور مطالعه تنوع ژنتیکی تومور و سیر تکاملی آن انجام شده است. دستاوردهای این تلاشها برای سرطان پستان (12–5)، سرطان تخمدان (13)، سرطان کولورکتال (15، 14)، سرطان ریه (16)، سرطان کبد (17)، سرطان کلیه (18)، سرطان سر و گردن (19)، ملانوم (20)، لوسمی میلوئیدی حاد (AML) (22، 21) و …، قابلتوجه بوده است. در جدول 1، پیشرفتهای اخیر در تحقیق ژنومیک سرطان با بکارگیری فنآوریهای NGS خلاصه شده است.
Table 1 Recent NGS-based studies in cancer
| Cancer | Experiment Design | Description | ref |
| Colon cancer | 72 WES, 68 RNA-seq, 2 WGS | Identify multiple gene fusions such as RSPO2 and RSPO3 from RNA-seq that may function in tumorigenesis | [15] |
| Breast cancer | 65 WGS/WES, 80 RNA-seq | 36% of the mutations found in the study were expressed. Identify the abundance of clonal frequencies in an epithelial tumor subtype | [11] |
| Hepatocellular carcinoma | 1 WGS, 1 WES | Identify TSC1 nonsense substitution in subpopulation of tumor
cells, intra-tumor heterogeneity, several chromosomal rearrangements, and patterns in somatic substitutions |
[17] |
| Breast cancer | 510 WES | Identify two novel protein-expression-defined subgroups and novel subtype-associated mutations | [5] |
| Colon and rectal cancer | 224 WES, 97 WGS | 24 genes were found to be significantly mutated in both cancers. Similar patterns in genomic alterations were found in colon and rectum cancers | [14] |
| squamous cell lung cancer | 178 WES, 19 WGS, 178
RNA-seq, 158 miRNA-seq |
Identify significantly altered pathways including NFE2L2 and KEAP1 and potential therapeutic targets | [16] |
| Ovarian carcinoma | 316 WES | Discover that most high-grade serous ovarian cancer contain TP53 mutations and recurrent somatic mutations in 9 genes | [13] |
| Melanoma | 25 WGS | Identify a significantly mutated gene, PREX2 and obtain a comprehensive genomic view of melanoma | [20] |
| Acute myeloid leukemia | 8 WGS | Identify mutations in relapsed genome and compare it to primary tumor. Discover two major clonal evolution patterns | [21] |
| Breast cancer | 24 WGS | Highlights the diversity of somatic rearrangements and analyzes rearrangement patterns related to DNA maintenance | [8] |
| Breast cancer | 31 WES, 46 WGS | Identify eighteen significant mutated genes and correlate clinical featuresofoestrogen-receptor-positivebreastcancerwithsomatic alterations | [7] |
| Breast cancer | 103 WES, 17 WGS | Identify recurrent mutation in CBFB transcription factor gene and deletion of RUNX1. Also found recurrent MAGI3-AKT3 fusion in triple-negative breast cancer | [6] |
| Breast cancer | 100 WES | Identify somatic copy number changes and mutations in the coding exons. Found new driver mutations in a few cancer genes | [9] |
| Acute myeloid leukemia | 24 WGS | Discover that most mutations in AML genomes are caused by random events in hematopoietic stem/progenitor cells and not by aninitiating mutation | [22] |
| Breast cancer | 21 WGS | Depict the life history of breast cancer using algorithms and sequencing technologies to analyze subclonal diversification | [12] |
| Head and neck squamous cell carcinoma | 32 WES | Identify mutation in NOTCH1 that may function as an oncogene | [19] |
| Renal carcinoma | 30 WES | Examine intra-tumor heterogeneity reveal branch evolutionary tumor growth | [18] |
کشف ژنهای جدید مرتبط با سرطان
سرطان بهطور عمده در اثر تجمع تغییرات ژنتیکی ایجاد میشود که ممکن است از طریق وراثتی در ردهی زایا و یا در طی دوره زندگی سلول، بهصورت اکتسابی ایجاد شود. اثرات این تغییرات بر روی انکوژنها، ژنهای سرکوبکننده تومور و یا ژنهای ترمیمکننده DNA، سبب میشود تا سلولها از مکانیسمهای نظارت بر رشد سلولی فرار کنند و بدون کنترل تکثیر یابند و تومور را بوجود آورند (23). اعقاب سلول سرطانی نیز ممکن است جهشهای بیشتری را تجربه کنند که منجر به گسترش کلونی میشود (24). همگام با گسترش کلونها، سلولها نیز توانائی تهاجم به بافتهای مجاور و بدنبال آن، ایجاد متاستاز را کسب میکنند (25).
تعیین توالی ژنومهای سرطانی قادر به شناسایی تعداد جدیدی از ژنهای مرتبط با سرطان، بهویژه سرطان پستان شده است. اخیراً شش مقاله، یافتههای خود را در مورد بررسیهای انجامشده بر روی سرطان پستان، بدین شرح گزارش کردهاند:
TCGA با تعیین توالی اگزونها روی 510 نمونه از 507 بیمار (5)، Banerji و همکاران روی 103 نمونه، تعیین توالی اگزونها و بر روی 17 نمونه تعیین توالی کل ژنوم را انجام دادند، Ellis و همکاران همین کار را به ترتیب روی 31 نمونه و 46 نمونه انجام دادند (7)، Stephens و همکارانش توالی اگزونها را روی 100 نمونه انجام دادند، Shah و همکارانش توالی کل ژنوم/ اگزون و RNA را روی 65 نمونه و 80 نمونه سرطان پستان که مارکرهای سهگانه منفی داشتند، کار کردند (11) و Nik – Zainal و همکارانش توالی کل ژنوم را روی 21 جفت تومور/ نرمال انجام دادند (12).
علاوه بر تأیید موتاسیونهای سوماتیک در ژنهای GATA3,TP53 و PIK3CA، این مطالعات، موتاسیونهای جدیدی را در ژنهای مرتبط با سرطان نیز کشف کردند. گرچه موتاسیونهای جدید بسیار کم اتفاق میافتند (کمتر از 10 درصد)، بااینحال آنها در زیرردهی سرطانهای پستان یافت شدند و میتوان آنها را در مسیرهای سیگنالینگ مرتبط با ایجاد سرطان قرار داد؛ بهعنوان مثال موتاسیونهای MAP3K1 در زیرگروه لومینال A بهکرات رخ میدهند (7،5). مسیرهای سیگنالینگ که دربرگیرنده P53، واسطههای تغییردهندهی ساختار کروماتین و مسیر ERBBمیباشند، بهکرات حامل جهشهای جدید بودهاند (11). علاوه بر این، برخی جهشها فرصتهای مناسب درمانی را بهصورت هدفمند فراهم میسازند، برای مثال جهشهای GATA3 بهعنوان یک مارکر، استفاده از ترکیبات مهارکننده آروماتاز را برای یک روش درمانی مطرح میسازد (7). همچنین، تعیین توالی ژنومیک به تشخیص و تعیین پروفایل مشخصات جهشها در سرطان کولورکتال کمک کرده است، مثلاً، تعیین توالی اگزوم انجام شده بر روی 72 جفت تومور- نرمال، 36303 جهشهای سوماتیک تغییردهندهی عملکرد پروتئین را شناسایی کرد. تجزیه و تحلیل بیشتر، منجر به شناسایی 23 کاندید ژنی شد که از آن بین میتوان به ژنهای KRAS ,TP53 و PIK3CA که در ایجاد سرطان بهخوبی شناخته شدهاند و همچنین ژنهای جدید همانند ATM که در کنترل سیکل سلولی دخیل هستند، اشاره کرد. تعیین توالی RNA منجر به تعیین ترکیبات پروتئینی موسوم به RSPO شده است که بهعنوان یک تنظیمکننده مهم مسیر سیگنالینگ Wnt قلمداد میشود و میتواند مسیرهای منتهی به تومورزائی را فعال کند (15). مثال دیگر، توالییابی اگزومی میباشد که بر روی 224 جفت تومور و نرمال انجام شده است. در این مطالعه، 15 ژن را در سرطانهای با فرکانس بالای جهش و 17 ژن را در سرطانهای با فراوانی پایین جهش، شناسایی کردند. در میان سرطانهای با فراوانی جهش پایینتر، جهشهای جدید در SOX9 ,ARID1A ,ATM و FAM123B در کنارجهشهای APC ,TP53 و KRAS که از قبل مطرح بودند، شناسایی شدند. تجزیهوتحلیل جهشهای SOX9,ARID1A,ATM و FAM123B و بررسی عملکرد آنها حاکی از این است که این ژنها با احتمال بالا با سرطان کولورکتال مرتبط هستند. سرطانهای با فرکانس پایین جهش کولون و رکتوم نیز از الگوی مشابهی تبعیت میکنند. تعیین توالی کامل ژنوم 97 تومور و نمونههای نرمال، همجوشی ژنهای NAV2 – TCF7L1 را مشخص کرد (14).
تکامل و ناهمگنی تومور
آنچه باعث میشود سرطان را نتوان بهراحتی کنترل نمود، ناپایداری ژنومی است که در سیر بیماری سرطان رخ میدهد که بهنوبهی خود باعث تنوع ژنتیکی در سلولهای سرطانی و سپس انتخاب و تکامل توده سرطانی میشود (26). این ایده، اولین بار در سال 1976 توسط پیتر نوول بهعنوان الگوی تکامل کلونی سرطان مطرح شد. کارهای بعدی که توسط سایر پژوهشگران که در دهه 1980 انجام شد، این نظریه را با مطالعات سابکلونهای متاستازدهنده یافتهی سللاین سارکوم موش، مورد تأیید قرار داد (26). استفاده گسترده از NGS منجر به ایجاد بینشی بنیادی به ناهمگنی و تکامل تومور شده است. تغییرات بین تومورها را ناهمگنی اینترتومور و تغییرات داخل یک تومور را ناهمگنی اینتراتومور مینامند. ناهمگنی بین توموری باعث ایجاد تفاوتهای ظاهری، تفاوت در الگوی بیان ژن و اختلاف در تعداد کپیهای مناطق تکراری میگردد که منجر به ایجاد زیرگروههای مختلف تودهی سرطانی میشوند (31-27). مطالعات TCGA و Eillis حاکی از آن است که اختلاف در الگوهای بیان ژن بدنبال جهشهای سوماتیک به وقوع میپیوندد (5 و 7).
تعیین توالیهای انجام شده توسط NGS، مبین این مطلب بود که هر تومور، دارای جهشهای خاص خود است و از این لحاظ منحصربهفرد میباشد، بهعنوان مثالStephens و همکارانش دریافتند که امکان ایجاد 73 ترکیب متفاوت ژنهای سرطانی جهشیافته در بین 100 سرطان پستان وجود دارد (9). ناهمگنی اینتراتومور را میتوان بهعنوان کلونهای سلولی مجزا یا سابکلونهایی درون یک تومور قلمداد کرد که نشاندهندهی خصوصیات مختلف، بافتشناسی مختلف، بیان ژن، قابلیت ایجاد متاستاز و تکثیر، میباشند. توانایی تولید دادههای با وضوح بالا، NGS را به ابزاری بسیار مفید برای مطالعه ناهمگنی اینتراتومورتبدیل کرده است. مطالعه اخیر مبتنی بر NGS بر روی کارسینوم سلول کلیوی از چهار بیمار، موفق به روشن شدن ناهمگنی اینتراتومور شده است (18). برای بیمار اول، تومور اولیه و نوع متاستاز شده به دیواره قفسه سینه از طریق بررسی اگزونی مورد ارزیابی قرار گرفت. از 128 موتاسیون تأیید گزارش شده در 9 ناحیه تومور اولیه، 40 مورد آن در همه مناطق تومور گزارش شد، 59 مورد، با برخی نواحی مشترک بود و 29 مورد منحصر به نواحی خاص بود که نشاندهندهی وجود ناهمگنی ژنتیکی درون تومور و تکامل کلونی ناحیهای میباشد (18). مهمتر از همه، مطالعات نشان داد که یک نمونه بیوپسی از تومور فقط منعکسکننده بخشی از جهشهای داخل تومور میباشد. به کمک یک نمونه بیوپسی، حدود 55 درصد جهشهای مربوط به تومور شناسایی شده که 34 درصد آنها در بیشتر نواحی تومور مشترک بود. ایجاد جهشهای داخل تومور به شکل مستمر باعث ایجاد ناهمگونی داخل بافت توموری میشود. بهعنوان مثال، مطالعات انجام شده بر روی کارسینوم سلول کلیوی و سرطان پستان مبین ساختار انشعابی در داخل تومور میباشد (18) و حاکی از این است که سلولهای سرطانی با گذشت زمان و تجمع جهشها، توانائیهای خاصی را بدست میآورند و با تشکیل کلونهای متفاوت، تکامل پیدا میکنند (26). مطابق فرضیه تنه- شاخه (26)، در ابتدا جهشهای سوماتیکی باعث رشد توده سرطانی شده است؛ این نوع جهشها در ابتدای تشکیل تومور، در بین سلولهای سرطانی مشترک میباشد، سپس با گذشت زمان، جهشهای سوماتیکی بیشتر باعث ناهمگنی شده و سابکلونها را تشکیل دادهاند که میتوان آنها را در تومورها و مناطقی که حاوی متاستاز هستند، یافت. در ادامه از بین سلولهای سرطانی متفاوت، تنها تعداد اندکی از سلولها توانایی تطبیق با شرایط را کسب کرده “Bottleneck Effect” و ایجاد سابکلونهای جدیدی را میکنند که میتواند به ناپایداری کروموزومی نیز منجر شود (26). سلولهای سرطانی که توانستهاند خود را با شرایط جدید محیط وفق دهند (این شرایط محیطی جدید میتواند مواجهه با یک دارو باشد)، به شکل بهتری رشد کرده و شرایط جدید را تحمل میکنند (مقاومت دارویی) (18)، لذا بهمنظور اقدام درمانی مناسب، بایستی جهشهایی را که در همه کلونها مشترک هستند و باصطلاح برروی تنه توده توموری قرار گرفتهاند، شناسایی کرد تا بتوان درمانهای دارویی را با اثردهی بهتر و راندمان بالاتر بکار گرفت.
در ترجمه این متن از راهنماییهای ارزنده همکار گرامی جناب آقای دکتر شهرام نعمتی برخوردار شدم که بدین وسیله مراتب قدردانی خود را از ایشان ابراز مینمایم.
References:
- Taylor BS, Ladanyi M: Clinical cancer genomics: how soon is now? J Pathol
2011, 223:318–326.
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE,
Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, Puzanov I, Lewis KD, Amaravadi RK,
Chmielowski B, Lawrence HJ, Shyr Y, Ye F, Li J, Nolop KB, Lee RJ, Joe AK, Ribas A: Survival in BRAF
V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012, 366:707–714.
- Metzker ML: Sequencing technologies – the next generation. Nat Rev Genet 2010, 11:31–46.
- Wold B, Myers RM: Sequence census methods for functional genomics.
Nat Methods 2008, 5:19–21.
- Cancer Genome Atlas Research Network: Comprehensive molecular portraits of human breast
tumours. Nature 2012, 490:61–70.
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS,
Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez- Lopez JC, Peng S, Ardlie KG, Auclair D,
Bautista-Pina V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH,
Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher
SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC,
Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J,
Toker A, Getz G, Hidalgo-Miranda A, Meyerson M: Sequence analysis of mutations and translocations
across breast cancer subtypes. Nature 2012, 486:405–409.
- Ellis MJ, et al: Whole-genome analysis informs breast cancer response to aromatase inhibition.
Nature 2012, 486:353–360.
- Stephens PJ, et al: Complex landscapes of somatic rearrangement in human breast cancer
genomes. Nature 2009, 462:1005–1010.
- Stephens PJ, et al: The landscape of cancer genes and mutational processes in breast cancer.
Nature 2012, 486:400–404.
- Nik-Zainal S, et al: The life history of 21 breast cancers. Cell 2012,
149:994–1007.
- Shah SP, et al: The clonal and mutational evolution spectrum of primary triple-negative breast
cancers. Nature 2012, 486:395–399.
- Nik-Zainal S, et al: Mutational processes molding the genomes of 21 breast cancers. Cell 2012,
149:979–993.
- Cancer Genome Atlas Research Network: Integrated genomic analyses of ovarian carcinoma.
Nature 2011, 474:609–615.
- Cancer Genome Atlas Research Network: Comprehensive molecular characterization of human colon
and rectal cancer. Nature 2012, 487:330–337.
- Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y,
Janakiraman V, Jaiswal BS, Guillory J, Ha C, Dijkgraaf GJ, Stinson J, Gnad F, Huntley MA,
Degenhardt JD, Haverty PM, Bourgon R, Wang W, Koeppen H, Gentleman R, Starr TK, Zhang Z,
Largaespada DA, Wu TD, de Sauvage FJ: Recurrent R-spondin fusions in colon cancer.
Nature 2012, 488:660–664.
- Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, Chu A, Collisson EA, Cope L, Creighton
CJ, Getz G, Herman JG, Johnson BE, Kucherlapati R, Ladanyi M, Maher CA, Robertson G, Sander C, Shen
R, Sinha R, Sivachenko A, Thomas RK, Travis WD, Tsao MS, Weinstein JN, Wigle DA, Baylin SB,
Govindan R, Meyerson M: Comprehensive genomic characterization of squamous cell lung cancers.
Nature 2012, 489:519–525.
- Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H,
Shirakihara T, Sakamoto H, Wang L, Ojima H, Shimada K, Kosuge T, Okusaka T, Kato K, Kusuda J,
Yoshida T, Aburatani H, Shibata T: High-resolution characterization of a hepatocellular carcinoma
genome. Nat Genet 2011, 43:464–469.
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N,
Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K,
Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M,
Szallasi Z, Downward J, Futreal PA, Swanton C: Intratumor heterogeneity and branched evolution
revealed by multiregion sequencing. N Engl J Med 2012, 366:883–892.
- Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J,
Wang J, Zhang N, El-Naggar AK, Jasser SA, Weinstein JN, Trevino L, Drummond JA, Muzny DM, Wu Y,
Wood LD, Hruban RH, Westra WH, Koch WM, Califano JA, Gibbs RA, Sidransky D, Vogelstein B,
Velculescu VE, Papadopoulos N, Wheeler DA, Kinzler KW, Myers JN: Exome sequencing of head and neck
squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333:1154–1157.
- Berger MF, et al: Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012,
485:502–506.
- Ding L, et al: Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome
sequencing. Nature 2012, 481:506–510.
- Welch JS, et al: The origin and evolution of mutations in acute myeloid leukemia. Cell 2012,
150:264–278.
- Wong KM, Hudson TJ, McPherson JD: Unraveling the genetics of cancer: genome sequencing and
beyond. Annu Rev Genomics Hum Genet 2011, 12:407–430.
- Cahill DP, Kinzler KW, Vogelstein B, Lengauer C: Genetic instability and darwinian selection in
tumours. Trends Cell Biol 1999, 9:M57–M60.
- Brosnan JA, Iacobuzio-Donahue CA: A new branch on the tree: next- generation sequencing in the
study of cancer evolution. Semin Cell Dev Biol 2012, 23:237–242.
- Swanton C: Intratumor heterogeneity: evolution through space and time.
Cancer Res 2012, 72:4875–4882.
- Russnes HG, Navin N, Hicks J, Borresen-Dale AL: Insight into the heterogeneity of breast cancer
through next-generation sequencing. J Clin Invest 2011, 121:3810–3818.
- Samuel N, Hudson TJ: Translating Genomics to the Clinic. Clinical chemistry: Implications of
Cancer Heterogeneity; 2012.
- Almendro V, Fuster G: Heterogeneity of breast cancer: etiology and clinical relevance. Clinical
& translational oncology: official publication of theShyr and Liu Biological Procedures Online 2013, 15:4
Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico 2011,
13:767–773.
- Yancovitz M, Litterman A, Yoon J, Ng E, Shapiro RL, Berman RS, Pavlick AC, Darvishian F,
Christos P, Mazumdar M, Osman I, Polsky D: Intra- and inter- tumor heterogeneity of
BRAF(V600E))mutations in primary and metastatic melanoma. PLoS One 2012, 7:e29336.
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa
S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerod A, Green A,
Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A,
Borresen-Dale AL, Brenton JD, Tavare S, Caldas C, Aparicio S: The genomic and transcriptomic
architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486:346–352.
- Desai AN, Jere A: Next-generation sequencing: ready for the clinics? Clin Genet 2012,
81:503–510.
- Welch JS, Westervelt P, Ding L, Larson DE, Klco JM, Kulkarni S, Wallis J, Chen K, Payton JE,
Fulton RS, Veizer J, Schmidt H, Vickery TL, Heath S, Watson MA, Tomasson MH, Link DC, Graubert TA,
DiPersio JF, Mardis ER, Ley TJ, Wilson RK: Use of whole-genome sequencing to diagnose a cryptic
fusion oncogene. JAMA 2011, 305:1577–1584.
- Li H, Ruan J, Durbin R: Mapping short DNA sequencing reads and calling variants using mapping
quality scores. Genome Res 2008, 18:1851–1858.
- Li H, Durbin R: Fast and accurate short read alignment with Burrows- Wheeler transform.
Bioinformatics 2009, 25:1754–1760.
- Li H, Durbin R: Fast and accurate long-read alignment with Burrows- Wheeler transform.
Bioinformatics 2010, 26:589–595.
- Langmead B, Salzberg SL: Fast gapped-read alignment with Bowtie 2. Nat Methods 2012,
9:357–359.
- Homer N, Merriman B, Nelson SF: BFAST: an alignment tool for large scale genome resequencing.
PLoS One 2009, 4:e7767.
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J: SOAP2: an improved ultrafast tool for
short read alignment. Bioinformatics 2009, 25:1966–1967.
- Ning Z, Cox AJ, Mullikin JC: SSAHA: a fast search method for large DNA databases. Genome Res
2001, 11:1725–1729.
- Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M: SHRiMP: accurate mapping of short
color-space reads. PLoS Comput Biol 2009, 5:e1000386.
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G,
Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB,
Altshuler D, Daly MJ: A framework for variation discovery and genotyping using next-generation DNA
sequencing data. Nat Genet 2011, 43:491–498.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The
Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25:2078–2079.
- Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K: SNP detection for massively parallel
whole-genome resequencing. Genome Res 2009, 19:1124–1132.
- Goya R, Sun MG, Morin RD, Leung G, Ha G, Wiegand KC, Senz J, Crisan A, Marra MA, Hirst M,
Huntsman D, Murphy KP, Aparicio S, Shah SP: SNVMix: predicting single nucleotide variants from
next-generation sequencing of tumors. Bioinformatics 2010, 26:730–736.
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding
L: VarScan: variant detection in massively parallel sequencing of individual and pooled samples.
Bioinformatics 2009, 25:2283–2285.
- Lam HY, Pan C, Clark MJ, Lacroute P, Chen R, Haraksingh R, O’Huallachain M, Gerstein MB, Kidd
JM, Bustamante CD, Snyder M: Detecting and annotating genetic variations using the HugeSeq
pipeline. Nat Biotechnol 2012, 30:226–229.
- Liu Q, Guo Y, Li J, Long J, Zhang B, Shyr Y: Steps to ensure accuracy in genotype and SNP
calling from Illumina sequencing data. BMC Genomics 2012, 13:S8.
- Wang W, Wei Z, Lam TW, Wang J: Next generation sequencing has lower sequence coverage and
poorer SNP-detection capability in the regulatory regions. Sci Rep 2011, 1:55.
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L,
Wilson RK: VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome
sequencing. Genome Res 2012, 22:568–576.
- Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK,
Ding L: SomaticSniper: identification of somatic point mutations in whole genome sequencing data.
Bioinformatics 2012, 28:311–317.
- Roth A, Ding J, Morin R, Crisan A, Ha G, Giuliany R, Bashashati A, Hirst M, Turashvili G,
Oloumi A, Marra MA, Aparicio S, Shah SP: JointSNVMix: a probabilistic model for accurate detection
of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 2012,
28:907–913.
- Kumar P, Henikoff S, Ng PC: Predicting the effects of coding non- synonymous variants on
protein function using the SIFT algorithm. Nat Protoc 2009, 4:1073–1081.
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev
SR: A method and server for predicting damaging missense mutations. Nat Methods 2010, 7:248–249.
- Wong WC, Kim D, Carter H, Diekhans M, Ryan MC, Karchin R: CHASM and SNVBox: toolkit for
detecting biologically important single nucleotide mutations in cancer. Bioinformatics 2011,
27:2147–2148.
- Wang K, Li M, Hakonarson H: ANNOVAR: functional annotation of genetic variants from
high-throughput sequencing data. Nucleic Acids Res 2010, 38:e164.
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q,
Locke DP, Shi X, Fulton RS, Ley TJ, Wilson RK, Ding L, Mardis ER: BreakDancer: an algorithm for
high-resolution mapping of genomic structural variation. Nat Methods 2009, 6:677–681.
- Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, Eichler EE, Sahinalp SC:
Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery.
Bioinformatics 2010, 26:i350–i357.
- Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang Z, Snyder M, Gerstein MB: PEMer: a
computational framework with simulation-based error models for inferring genomic structural
variants from massive paired-end sequencing data. Genome Biol 2009, 10:R23.
- Zeitouni B, Boeva V, Janoueix-Lerosey I, Loeillet S, Legoix-ne P, Nicolas A, Delattre O,
Barillot E: SVDetect: a tool to identify genomic structural variations from paired-end and
mate-pair sequencing data. Bioinformatics 2010, 26:1895–1896.
- Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq.
Bioinformatics 2009, 25:1105–1111.
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou
CM, MacLeod JN, Chiang DY, Prins JF, Liu J: MapSplice: accurate mapping of RNA-seq reads for splice
junction discovery. Nucleic Acids Res 2010, 38:e178.
- Au KF, Jiang H, Lin L, Xing Y, Wong WH: Detection of splice junctions from paired-end RNA-seq
data by SpliceMap. Nucleic Acids Res 2010, 38:4570–4578.
- Wu TD, Nacu S: Fast and SNP-tolerant detection of complex variants and splicing in short
reads. Bioinformatics 2010, 26:873–881.
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras
TR: STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29:15–21.
- Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol 2010,
11:R106.
- Robinson MD, McCarthy DJ, Smyth GK: edgeR: a Bioconductor package for differential expression
analysis of digital gene expression data. Bioinformatics 2010, 26:139–140.
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L: Differential analysis of
gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 2012, 31:46–53.
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL,
Pachter L: Differential gene and transcript expression analysis of RNA-seq experiments with TopHat
and Cufflinks. Nat Protoc 2012, 7:562–578.
- Griffith M, Griffith OL, Mwenifumbo J, Goya R, Morrissy AS, Morin RD, Corbett R, Tang MJ, Hou
YC, Pugh TJ, Robertson G, Chittaranjan S, Ally A, Asano JK, Chan SY, Li HI, McDonald H, Teague K,
Zhao Y, Zeng T, Delaney A, Hirst M, Morin GB, Jones SJ, Tai IT, Marra MA: Alternative expression
analysis by RNA sequencing. Nat Methods 2010, 7:843–847.
- Katz Y, Wang ET, Airoldi EM, Burge CB: Analysis and design of RNA sequencing experiments for
identifying isoform regulation. Nat Methods 2010, 7:1009–1015.
- Kim D, Salzberg SL: TopHat-Fusion: an algorithm for discovery of novel fusion transcripts.
Genome Biol 2011, 12:R72.
- Chen K, Wallis JW, Kandoth C, Kalicki-Veizer JM, Mungall KL, Mungall AJ, Jones SJ, Marra MA,
Ley TJ, Mardis ER, Wilson RK, Weinstein JN, Ding L: BreakFusion: targeted assembly-based
identification of gene fusions in whole transcriptome paired-end sequencing data. Bioinformatics
2012, 28:1923–1924.
- Li Y, Chien J, Smith DI, Ma J: FusionHunter: identifying fusion transcripts in cancer using
paired-end RNA-seq. Bioinformatics 2011, 27:1708–1710.
- McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A,
Senz J, Melnyk N, Pacheco M, Marra MA, Hirst M, Nielsen TO, Sahinalp SC, Huntsman D, Shah SP:
deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol 2011,
7:e1001138.
- Piazza R, Pirola A, Spinelli R, Valletta S, Redaelli S, Magistroni V, Gambacorti- Passerini C:
FusionAnalyser: a new graphical, event-driven tool for fusion rearrangements discovery. Nucleic
Acids Res 2012, 40:e123.
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM: Inference of
patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM.
Bioinformatics 2010, 26:i237–i245.
- Cerami E, Demir E, Schultz N, Taylor BS, Sander C: Automated network analysis identifies core
pathways in glioblastoma. PLoS One 2010, 5:e8918.
- Ciriello G, Cerami E, Sander C, Schultz N: Mutual exclusivity analysis identifies oncogenic
network modules. Genome Res 2012, 22:398–406.
- Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E,
Garraway LA, Pe’er D: An integrated approach to uncover drivers of cancer. Cell 2010,
143:1005–1017.
- Langmead B, Hansen KD, Leek JT: Cloud-scale RNA-sequencing differential expression analysis
with Myrna. Genome Biol 2010, 11:R83.
- Anders S, Reyes A, Huber W: Detecting differential usage of exons from RNA-seq data. Genome Res
2012, 22:2008–2017.
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A,
Teague JW, Campbell PJ, Stratton MR, Futreal PA: COSMIC: mining complete cancer genomes in the
Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011, 39:D945–D950.
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML,
Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N: The cBio cancer genomics portal: an
open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012, 2:401–404.
- Gundem G, Perez-Llamas C, Jene-Sanz A, Kedzierska A, Islam A, Deu-Pons J, Furney SJ,
Lopez-Bigas N: IntOGen: integration and data mining of multidimensional oncogenomic data. Nat
Methods 2010, 7:92–93.
- Baudis M, Cleary ML: Progenetix.net: an online repository for molecular cytogenetic aberration
data. Bioinformatics 2001, 17:1228–1229.
- Treangen TJ, Salzberg SL: Repetitive DNA and next-generation sequencing: computational
challenges and solutions. Nat Rev Genet 2012, 13:36–46.
- Cooper GM, Shendure J: Needles in stacks of needles: finding disease- causal variants in a
wealth of genomic data. Nat Rev Genet 2011, 12:628–640.
- Nekrutenko A, Taylor J: Next-generation sequencing data interpretation: enhancing
reproducibility and accessibility. Nat Rev Genet 2012, 13:667–672.
- Eisenstein M: Reading cancer’s blueprint. Nat Biotechnol 2012, 30:581–584.
- Katsios C, Papaloukas C, Tzaphlidou M, Roukos DH: Next-generation sequencing-based testing for
cancer mutational landscape diversity: clinical implications? Expert Rev Mol Diagn 2012, 12:667–670
[1]Next Generation Sequencing
ژنومیکس و کاربرد آن در تشخیص بیماریها (2)
تست غیرتهاجمی تشخیص پیش از تولد
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام