
پلیمورفیسمهای تکنوکلئوتیدی در درمان و تحقیقات سرطان
شیماسادات سیدموسوی1، سمانه دولتآبادی2
1: دانشجوی کارشناسی ارشد ژنتیک، گروه زیستشناسی، دانشگاه آزاد اسلامی، نیشابور، ایران
2: استادیار، گروه زیستشناسی، دانشگاه آزاد اسلامی، نیشابور، ایران
نویسنده مسئول: شیماسادات سیدموسوی، آدرس ایمیل: shimamosavi785@gmail.com
چکیده
چندشکلیهای تکنوکلئوتیدی[1] (SNP)، تفاوت یافتشده در یک نوکلئوتید نسبت به موقعیت مشابه آن در توالی DNA است. SNPها تغییرات و تنوعهای توالی طبیعی با دانسیته بالا در ژنومها هستند.SNP ها بهعنوان یک منبع ژنتیکی عمده از تغییر فنوتیپی درون یک گونه در نظر گرفته میشوند و مارکر ژنتیکی مهم و خوبی بهحساب میآیند. SNPها کاربردهای فراوانی از جمله بهعنوان نشانگر ملکولی در تحقیقات ژنتیکی و اصلاح، بهعنوان نشانگر ملکولی در مطالعات ژنتیکی بیماریها و ژنومیکس دارو، در نقشهیابی ژنتیکی، انتخاب به کمک نشانگر و … دارند. SNPها در طول توالي ژنهای مرتبط با خطر ابتلا به انواع سرطانها طی سالهای اخیر بسیار بررسیشدهاند. در این مطالعه قصد داریم مطالبی را در ارتباط با نقش SNP در خطر ابتلا به انواع سرطان بهطور خلاصه مکتوب کنیم.
مقدمه
پلیمورفیسم تک نوکلئوتید به انگلیسی بهطور مخفف SNP ؛ یک تغییر در توالی DNA است که در آن یک نوکلئوتید A,C،G,T در ژنوم افراد یک گونه بیولوژیکی یا بین یک جفت کروموزوم در یک فرد فرق دارد. بهعنوان مثال، دو توالی از قطعات DNA از دو فرد متفاوت، AAGCCTA به AAGCTTA، در یک نوکلئوتید با هم متفاوتند. در این حالت گفته میشود که دو الل C و Tوجود دارند. تقریباً تمام SNPهای مشترک فقط دو الل دارند.
در یک جمعیت، میتوان یک فرکانس الل حداقلی[2] را به SNPها نسبت داد. فرکانس الل حداقلی یعنی کمترین فراوانی یک الل در یک لوکوس[3] که در جمعیت خاصی مشاهده شده است. فرکانس الل حداقلی کمتر از فرکانس دو الل برای پلیمورفیسم تک نوکلئوتیدی است. بین جمعیت انسانها تفاوتهای گوناگونی وجود دارد، بنابراین یک الل SNP که در یک منطقه جغرافیایی یا در یک گروه نژادی مشترک است، ممکن است بین گونههای دیگر کمتر باشد.
SNPی که در آن هر دو الل یک توالی یکسان از پلیپپتید را تولید میکنند، یک پلیمورفیسم مترادف[4] نامیده میشود. اگر یک دنباله پلیپپتید متفاوت تولید شود به آن پلیمورفیسم جایگذاری[5] میگویند. یک پلیمورفیسم جایگذاری میتواند دارای معنی اشتباه[6] باشد که در آن صورت منجر به ایجاد آمینواسید متفاوتی خواهد شد، یا اینکه میتواند بدون معنی[7] باشد که در این صورت منجر به یک توقف کدون زودرس[8] میشود. بیشتر از نصف امراض مرتبط با جهش، به علت پلیمورفیسمهای جایگذاری هستند. SNPهایی که در ناحیه کد پروتئین نیستند ممکن است همچنان روی اسپلایسینگ و نسخهبرداری ژن تأثیر بگذارند. بیانهایی از ژن که از این نوع SNP تأثیر پذیرفته باشند eSNP (expression SNP) نامیده میشوند. تغییراتی که در دنبالهی DNA در انسانها اتفاق میافتد میتواند روی توسعه امراض و جواب به پاتوژنها، مواد شیمیایی، داروها، واکسنها و دیگر عاملها تأثیر بگذارد (1).
مثالهایی از SNPها
- rs6311 و rs6313، SNPهایی در ژن گیرنده HTR2A سروتونین روی کروموزوم 13 در انسان هستند.
- SNPیی در ژن F5 که موجب ترومبوفیلیا در فاکتور 5 لیدین[9]میشود.
- rs3091244 مثالی از یک SNP تریآللیک[10] در ژن CRP روی کروموزوم شماره یک انسان است.
- کدهای TAS2R38 در قابلیت PTC tasting (phenylthiocarbamide) که شامل 6 SNP مشخص است.
- SNPهای rs148649884 و rs138055828 در ژن فیکولین 1 (FCN1) که فیکولین M را کد میکند و قابلیت اتصال لیگاند را در پروتئین فیکولین M نوترکیب مختل میکند (2).
تنوع ژنتیکی در ژنوم انسان
در فرایند دریفت توالی ژنوم انسان مشخص شده که محتوای تنوع ژنتیکی بیشتر از آن چیزی است که قبلاً تخمین زده شده بود (4-3). همانطور که گفته شد، رایجترین تنوع توالی در ژنوم انسانی جایگزینی پایدار یک تکباز یعنی پلیمورفیسم تکنوکلئوتیدی (SNP) است. SNP ها در یک جمعیت با فرکانس اللی حداقل بیشتر از 1 درصد هستند (5). اکثر SNPها خاموش هستند و عملکرد بیان ژن را تغییر نمیدهند. از نظر مفهومی، منطقی است که اصطلاح “جهش” برای تغییرات نادر با نفوذ بسیار بالا اختصاص داده شده است و معمولاً با یک فنوتیپ مضر مانند یک اختلال مونوژنیک کلاسیک (مثل کمخونی داسی شکل یا هموفیلی) همراه است. تعداد کل SNPها در ژنوم انسان بیشتر از 10 میلیون تخمین زده شده است (6) و تعداد SNPهای دارای فرکانس الل حداقلی بیش از 10 درصد بهنظر میرسد تا 5 میلیون باشد (7). SNPها در سرتاسر ژنوم انسانی و با فرکانس الل حداقلی 1 در هر 1000 جفت باز و با تفاوتهای منطقهای مشخص توزیع شدهاند (8). SNPها به دلیل موتاسیونهای نقطهای ایجاد میشوند که بهطور انتخابی در جمعیت حفظ میشوند. فراوانی SNPها به چهار فاکتور وابسته است:
1) میزان زمانی که صرف موتاسیون میشود،
2) فشار فرگشتی (تکاملی) بر روی تغییرات مشخص بیولوژیکی و آنهایی که در ارتباط با واریانت کاربردی هستند،
3) دریفت ژنتیکی تصادفی و
4) رویدادهای اتفاق افتاده در شرایط بد زیستی[11].
SNPها در نواحی کروموزومی مشابه بهصورت تصادفی به ارث نرسیدهاند بلکه بهعنوان ترکیبی از آللها هستند که بلوکهای هالوتایپ را تشکیل میدهند. بهنظر میرسد ژنومها درون بلوکهای متمایزی به نام LD سازماندهی شدهاند (9)، بنابراین، پیچیدگی آنالیز کردن SNPها در یک ژن یا لوکوس میتواند توسط آنالیز مارکرهای بهارثرسیده از یک هالوتایپ کاهش یابد. بهطور عملی، هالوتایپها میتوانند توسط آنالیز LD یک ناحیه در افراد غیرخویشاوند یافت شوند و یا بهصورت مولکولی در دودمان خانواده مشخص شوند (شکل 1). قابل توجه است که فراوانی هر دو یعنی SNPها و مقدار LD ممکن است بهطور قابلتوجهی بین جمعیتها متفاوت باشد. علاوه بر این، واریانتهای خاص جمعیتی[12] بسیار زیادی وجود دارد (8).
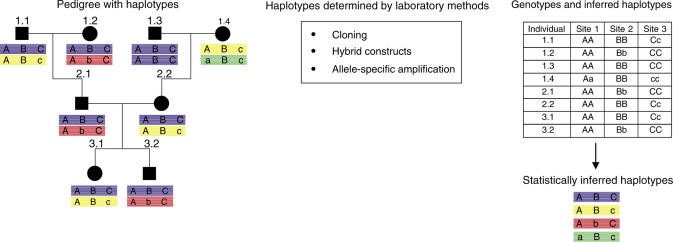
شکل 1: تعیین هالوتایپ در افراد خویشاوند و غیرخویشاوند
ساختار هالوتایپ در دودمان بر اساس آنالیز در 3 نقطهی خاص از ناحیه کروموزومی تعیین شده است. اللهای مینور و ماژور با اشکال A/a، B/b و C/c به ترتیب نشان داده شدهاند. هالوتایپ میتواند از اطلاعات جدول منتج شده باشد و مدل وراثت کلاسیک مندل را دنبال کند. هالوتایپها میتوانند از افراد غیرخویشاوند با بکارگیری الگوریتم آماری برای حدس زدن هالوتایپها بر اساس اطلاعات ژنوتایپ بکارگیری شوند. با این وجود، هالوتایپ را بهوضوح تعیین نخواهد کرد، در عوض هالوتایپی با احتمالات آماری صحیح را خواهد داد. هرچند این روش بسیار زیاد و بیشتر به خاطر هزینه بهرهوری پایین استفاده شده است، روشهای دیگری وجود دارند که هالوتایپ را به صورت قطعی تعیین میکنند. گاهی اوقات، شخص میتواند کلونهای DNA یک کروموزوم تک را برای آنالیز توالی مستقیم جدا کند و یا به طور گزینشی با استفاده از پرایمرهای الیگونوکلئوتیدهای خاص، آلل را تقویت کند.
SNP ها و فنوتایپ
به موازات اینکه سن علم ژنومیک برای جستجوی واریانتهای ژنتیکی (مانند SNPs) که بر حساسیت و پیامد بیماری اثر میگذارد، بیشتر میشود، تلاشهای زیادی در زمینه انتخاب SNPها برای مطالعه صورت گرفته است.
پیام نویدبخش استفاده از SNPهای دو آللی برای مطالعات ارتباط کلی ژنوم، هرچند بهصورت تئوری بسیار هیجانانگیز است، هنوز به دلیل مسائل مربوطه از جمله هزینههای هنگفت و غیرعملی بودن ژنوتایپینگ، هزاران SNP موردنیاز گاهی اوقات کنار گذاشته شده است، بهجز در توسعه پایگاههای دادهها و ابزار تحلیلی موردنیاز برای تفسیر دادهها (10)، با این حال ما میتوانیم در آینده به این رویکرد بپردازیم، اما در حال حاضر به دلیل منابع محدود و ابزارهای تحلیلی، اکثر محققان همچنان از استراتژیی استفاده میکنند که ژنهای خاصی را مورد بررسی قرار میدهد و شناخته شدهاند. روش ژن کاندید، شناسایی SNPها را در ژنهای دارای معنی مورد ارزیابی قرار میدهد؛ به عبارت دیگر، آنها با درک درستی از زیستشناسی در تناسب هستند. پلیمورفیسم تک نوکلئوتیدی همچنین میتواند از یک ناحیه ژنی که قبلاً توسط مطالعه ارتباط[13] شناسایی شده و یا بیان آن توسط روش میکروآرایه آنالیز شده، انتخاب شود. تلاشهای شدیدی بر روی SNPها متمرکز شده است که عملکرد پروتئین یا بیان ژن را تغییر میدهد. سلسله مراتبی نیز برای پیشبینی بیان یک فنوتیپ ممکن، برای SNPها پیشنهاد شده است (5). تخمین زده شده که احتمالاً 50000-250000 SNP وجود دارد که اثرات بیولوژیکی خاصی را بهوجود میآورند و اکثر آنها در 30000 ژن توزیع میشوند (5).
پیشبینی یک اثر بیولوژیکی احتمالاً پیچیدهتر از آن است که در اینجا بحث شود؛ بهعنوان مثال، یک SNP مترادف که توالی آمینواسید را تغییر نمیدهد، نشان داده است که بر پایداری نسخه DRD2 تأثیر میگذارد و منجر به تغییر بیان آن میشود (11). ازآنجاییکه اکثریت SNPها به تغییرات فنوتیپی منتج نمیشوند، بلکه متکی بر هالوتیپهای اجدادیند، یک تمایز اساسی بایستی بین SNPها بهعنوان نشانگرهای ژنتیکی و عوامل مرتبط با یک اثر فنوتیپی ایجاد شود.
تا به امروز، مکتوباتی (از جمله مقالات و کتب) که بر SNPها متمرکز شده، اثر کاربردی پیشبینیشده یا اثباتشده دارند. روش ژن کاندید که حاوی هاپلوتیپهای چسبیده به SNP است به بررسی تنوع ژنتیکی در ژن یا لوکوس میپردازد. هنگامی که یک هالوتایپ بهعنوان نشانگر برای یک فنوتیپ تأئید شده است، لازم است که SNP جزئی برای تعیین واریانتهای مسبب مورد بررسی قرار گیرد.
در برخی موارد لازم است SNPهای اضافی بهمنظور تعریف بهتر ساختار هالوتایپ در هنگام جستجوی واریانتهای مسبب مورد مطالعه قرار گیرند. تلاش برای کنار گذاشتن این و انتخاب فقط “SNPهای مهم عملی” فرصت را برای “علامتگذاری” یک ژن یا منطقه محدود میکند. علاوه بر این، انتظار میرود که جمعآوری SNP فردی، یک استراتژی مؤثر برای شناسایی تغییرات در ژنهایی باشد که میتواند به بیماریهای پیچیده مانند سرطان کمک کند. بدون تردید، مطالعات پیشین نشان میدهد که کدام واریانت برای مطالعه، پیشنهاد خواهد شد، هرچند پیشرفتهای فنی و بیوانفورماتیک در حال حاضر یک منبع غنی برای مطالعهاند که شامل بسیاری از ژنها و SNPها با شناخت کم میباشند.
موضوعات تحلیلی در مطالعات SNP
پیشبینی LDها و ساختار هالوتایپها، ابزاری قدرتمند برای اجرای مطالعات مربوطه ارائه میدهد (12). حتی اگر میزان LD و تعداد هالوتیپها در ژنوم و بین جمعیتها متفاوت باشد، به نظر میرسد که تنوع هالوتیپی کم برای هر لوکوس، همراه با برخی از تفاوتها در جمعیتهای جداگانه دیده شود (13)، در نتیجه، مطالعات مرتبط میتواند مجموعهی محدودی از SNPها را ژنوتایپ کند که به هالوتایپهای رایج، همراه با SNPهای برچسبگذاریشده توسط هالوتایپ (ht-SNPs) میانجامد (14). تجزیه و تحلیل با ht-SNPs موجب صرفهجویی مالی و DNA میشود، اما ابزارهای تجزیه و تحلیل جدید برای تخمین اثر هالوتایپهای فردی بر روی هر دو اثر اصلی (یعنی حساسیت یا پیامد[14]) و تعاملات ژن-ژن، موردنیاز است.
در حال حاضر مطالعات متعددی کاربرد تجزیه و تحلیل هالوتایپ را نشان دادهاند؛ بهعنوان مثال، ارتباطی بین هاپلوتایپها در کروموزوم 19 و کارسینوم سلول پایهای گزارش شده است. به همین ترتیب، هالوتیپهای پروموتور اینترلوکین 4 با کاندیدیازیس منتشره مزمن؛ یک عفونت خطرناک در بیماران مبتلا به لوسمی حاد، همراه است (15-16).
مطالعات مربوطه به خاطر عدم توانایی بکارگیری دائم نتایج تجدیدپذیر بسیار سخت است. تعدادی از عوامل به این ابهام کمک میکنند و در میان کارشناسان در این زمینه بحث بسیار میشود. از آنجا که تکرار برای پذیرش رابطه علتی بین SNP یا یک هالوتایپ و یک نتیجه حیاتی است، استدلال شده است که ارتباطات مثبت کاذب بهتر از منفی کاذب تحمل میشود. در حال حاضر، مشکل این است که اثرات کاذب بسیاری وجود دارد. برخی معتقدند که اجرای تعداد زیادی از تستها، به تعبیری، باعث افزایش میزان مثبت کاذب میشود، اما برخی دیگر معتقدند که متغیرها مستقل نیستند، بهخصوص زمانی که هالوتیپها مورد توجهاند (18-17).
یکی دیگر از عوامل عدم تجدیدپذیری، مطالعات بالقوه ناکافی است؛ چه در مطالعات اولیه و چه در مطالعات پس از آن. ترکیبی از جمعیتها بهعنوان عاملی گیجکننده پیش میروند، هرچند که تأثیر آن در نمونههای منتشرشده کمتر از پیشبینیشده است (19). با این حال، دریفت ژنتیکی همراه با نرخ و توزیع ژنومی وقایع نوترکیبی و جهش در جمعیتی که تحت فشارهای متنوعی قرار دارد، LD را تحت تأثیر قرار میدهد و به همین ترتیب، پتانسیلی برای کشف یک ارتباط داده شده است (18-17، 20). در حال حاضر، راهحلهای جایگزین پیشرفت کردهاند که در بردارنده تحلیل آماری جدید میباشند که به روشهای بیزی[15] و متاآنالیز مطالعات چاپشده میپردازد. پیشبینی شده است که ابزارهای تحلیلی جاری بهسرعت در واکنش به بازنگری مجموعه دادهها تکامل خواهند یافت.
SNP در تحقیقات سرطان
مطالعات ارتباط ژنتیکی با SNPها که سرطان را هدف قرار میدهند را میتوان به دو دسته گسترده، مطالعه حساسیت و پیامد تقسیم کرد (جدول 1).
پیامد، تعیین اطلاعات پیشآگهی، عوارض یا پاسخ به مداخلات دارویی را جستجو میکند (بهعنوان مثال، فارماکوژنومیکها). تا به امروز، تقریباً تمام مطالعات چاپشده، تعداد کمی از SNPها، ژنها و در موارد خاص واریانتهای مربوط به ژنهای یک مسیر یا فرایند بیولوژیکی را بررسی کردهاند (مانند آنزیمهای تعمیر DNA از قبیل XRCC1 و XRCC3 یا ژنهای متابولیسم زنوبیوتیک[16] مانند NAT1 و NAT2). اگرچه در ابتدا در مطالعه SNPها و سرطان، پیشرفتهای فنی و بیوانفورماتیک امکان افزایش تعداد ژنهای مورد استفاده در یک مطالعه را بهطور قابلتوجهی افزایش داده است، اما بسیار مهم است که توجه داشته باشیم مطالعات SNP قبل از پذیرش و قطعاً قبل از اجرای بالینی نیاز به تکرار دارند؛ بنابراین، مهم است که مکتوبات منتشرشده قبلی را بهعنوان تحلیلی بسیار اولیه از آنچه قطعاً یک فرایند پیچیده باشد، در نظر بگیریم.
جدول 1: مثالهایی از ژنها و ارتباط آنها
| ژن | ارتباط |
| حساسیت به سرطان | |
| میلوپروکسیداز[17]، MPO | سرطان ریه |
| ان-استیل ترانسفراز [18]1، NAT1 | سرطان مثانه |
| ان-استیل ترانسفراز، NAT2 | سرطان کولون و مثانه |
| پیامد سرطان | |
| CYP3A4 | سرطان پروستات |
| فارماکوژنومیکها | |
| تیوپورین[19]، اس-متیل ترانسفراز[20]، TPMT | سمیت هماتولوژیکی |
حساسیت به سرطان
احتمال ابتلا به یک سرطان خاص به احتمال زیاد با مجموعهای از انواع ژنتیکی مرتبط است که بسیاری از آنها میتوانند با عوامل محیطی در ارتباط باشند. تاکنون مطالعات اولیه، ژنهای منفردی از یک نمونه را ایجاد کرده که درنهایت تعامل ژن-ژن ها را میتواند بررسی کند. بهعنوان مثال، مطالعات در مورد سرطان ریه ژنهایی را که برای متابولیسم تنباکو و اعتیاد به نیکوتین مهم هستند را مورد بررسی قرار دادهاند. این رویکرد متمرکز بر روی برهمکنش یک محرک سرطانزای محیطی است و به دنبال شناسایی واریانتهای ژنتیکی است که حساسیت یا حفاظت در برابر دود تنباکو را تشخیص میدهند. در این رابطه، برهمکنش ژن و محیط، نشاندهنده استرس بر میزبان است و شاید اثر فنوتیپی SNP را تقویت کند. بهعنوان مثال، ژن میلوپرکسیداز؛ MPO، بهطور گسترده مورد مطالعه قرار گرفته است و دادههای تکرارپذیری را در مطالعات روی سفیدپوستان ارائه کرده است. نتیجه عملکرد انتقال G به A در 463- پروموتور پروگزیمال منجر به کاهش بیان mRNA MPO میشود. در افرادی که برای آلل A هموزیگوت هستند، خطر ابتلا به سرطان ریه نسبت به افرادی که دارای دو آلل G هستند، بهطور قابلتوجهی کمتر است (22-21).
یکی از قویترین موارد برای ارتباط پلیمورفیسم در ان- استیل ترانسفراز (NAT2) با سرطان مثانه و کولون است. NAT1 و NAT2 آنزیمهایی را کد میکنند که برای تبدیل آمینهای معطر و هتروسیکلیک مهم هستند و بهعنوان سرطانزا شناخته میشوند. خطر ابتلا به سرطان مثانه بهطور خاصی در فنوتیپ استیلاتور NAT2 بالا است و با سابقه مصرف سیگار افزایش مییابد. از سوی دیگر، برای سرطان کولون مرتبط با آمین هتروسیکلیک، فنوتیپ استیلاتور سریع NAT2 خطر بیشتری را به همراه دارد (24-23).
SNPها و پیامد
واریانتها میتوانند با پیامدها در ارتباط باشند و این میتواند در تصمیمگیری بالینی کمک کند؛ بهعنوان مثال واریانتهای ژنتیکی میتوانند خطر ابتلا به تومور متاستازدهنده یا تهاجمی را تغییر دهند. تا به امروز، مطالعات شفافی اهمیت واریانتهای ژرملاین[21] را بهعنوان نشانگرهای پیشآگهی نشان دادهاند، اما ازآنجاییکه درصد کمی از ژنهای شناختهشده به اندازه کافی مورد مطالعه قرار گرفتهاند، بررسی SNPها همچنان فعال باقی مانده است. تاکنون، نتایج اولیه نشان میدهد که SNPها در CYP3A4 با پیامد درازمدت سرطان پروستات در ارتباطند. پروموتور SNPی A-290G در CYP3A4 که یک ژن فعال اکسیداسیون تستوسترون به-B2، 6B، یا B- 15هیدروکسی تستوسترون است، به نظر میرسد که با شدت بیماری در ارتباط است. این اثر در مردان مسن بدون سابقه خانوادگی سرطان پروستات قویتر است (26-25).
چند مطالعه که ارتباط SNP و سرطان را نشان میدهد
SNP در جایگاههای هدف میکرو RNA حساسیت تومور را تحت تأثیر قرار میدهد
میکرو RNAها خانوادهای از RNAهای درونی، کوتاه و غیرکدشوندهاند که تنظیم ژن را بعد از فرایند رونویسی تعدیل میکنند. میکرو RNAها نقش تنظیمی خود را روی بیان ژن کدکننده پروتئین[22] (PCG) با اتصال کامل یا جزئی به منطقه ترجمه نشدهی 3Ꞌ (UTR) و همچنین درون توالی کدشده (CDS) و ناحیه ترجمهنشده 5Ꞌ مربوط به mRNA اعمال میکنند و درنهایت این فرایند دوام mRNA و ترجمه را تحت تأثیر قرار میدهد. نقش miRNA در پاتوژنزیز سرطان انسان، با شناسایی تغییرات ژنتیکی در لوکوس miRNA، سیگنیچرهای بیان miRNA که فنوتیپهای نئوپلاستیک مختلف را تعریف میکند، انکوژنهای بیشمار و ژنهای سرکوبگر تومور بهخوبی شناسایی شده است.
سرطان سینه یکی از شایعترین بدخیمیها در زنان است که هرسال بیش از 1 میلیون مورد جدید شناسایی میشود. بهطور گسترده در جهان پذیرفته شده که اکثر سرطانهای سینه بایستی محصولات ترکیبی آللهای کمنفوذ چندگانه[23] اختصاصی باشند. با این وجود، پیچیدگی واریانتهای شناساییشده بهتنهایی نمیتواند برای تقریباً 80 درصد موارد سرطان سینه خانوادگی که ارتباطی با ژنهای حساسیت پرنفوذ[24] سرطان سینه ندارند، بهحساب آید. نقش واریانتهای پلیمورفیک که در ژنهای مرتبط با سرطان سینه واقعند در ارتباط با حساسیت تومور بهطور گستردهای بیان شده است.اما هیچیک از گزارشهایی که مداخلات پاتوژنتیک تغییرات ژنومی را روی حساسیت سرطان سینه تحقیق میکنند، نقش SNPها را در تنظیم ژن miRNA: mRNA و اثر آن را در توسعه سرطان سینه ارزیابی نکردهاند. ملینا[25] و همکاران SNPهای مرتبط با حساسیت سرطان سینه برای تواناییشان در تحت تأثیر قرار دادن جایگاههای اتصال miRNA و تنظیم ژن miRNA: mRNAرا مورد تجزیه و تحلیل قرار دادند. ملینا و همکاران یک مکانیسم پاتوژنتیک جدید را شناسایی کردند تا ارتباط SNPهای خاص را با حساسیت سرطان سینه با استفاده از اطلاعات اخیر روی تنظیم ژن پس از ترجمه[26] توسط miRNA توضیح دهند (27).
SNP در پروموتور ماتریکس متالوپروتئیناز 1 و 3 به ترتیب حساسیت به سرطان ریه و سرطان سینه را افزایش میدهد
مثال غالب برای گسترش سرطان، یک پروسه چندمرحلهای پیچیده است که طی آن یک سلول نرمال تحت تغییرات ژنتیکی قرار میگیرد که منجر به تغییرات فنوتیپی و کسب توانایی هجوم و کلونیزه شدن در جایگاههای دورتر است. اگرچه بسیاری از فاکتورها در گسترش تومور درگیرند، برهمکنش بین سلولهای نئوپلاستیک و ریزمحیطهای احاطهکننده[27] برای هر گام از توموریژنزیز[28] حیاتی و ضروریاند (28).
یکی از پتانسیلهایی که نقش حیاتی را در پویایی حفظ میکرومحیطهای سلولی دارد، خانواده ماتریکس متالوپروتئیناز[29] است. خانواده ماتریکس متالوپروتئیناز شامل بیش از 20 آنزیم هستند که در ارتباط با تجزیهی غشای خارج سلولی[30] (ECM) از جمله غشای پایه میباشند. اختلال در یکپارچگی غشای پایه، شکلی از تومور تهاجمی، به تومور اجازه میدهد تا بهطور محلی و غیرمحلی پراکنده شود (29)؛ بنابراین این مسئله بهطور اولیه مورد اعتقاد است که ماتریکس متالوپروتئینازها از طریق تجزیه موانع فیزیکی در هجوم تومور، نفوذ به رگ خونی و متاستاز درگیر است. هرچند، علاوه بر تقویت هجوم سلولی توسط تخریب موانع غشای خارج سلولی، ماتریکس متالوپروتئینازها میتوانند همچنین میکرومحیطها را با استفاده از سیگنالهای سلولی تحت تأثیر قرار دهند (30). بیشتر ماتریکس متالوپروتئینازها نهتنها توسط سلولهای سرطانی بهصورت ژنتیکی تغییر یافتهاند، بلکه توسط سلولهای استرومال مجاور و مداخلهکننده سنتز میشوند (31).
نقش SNP در پروموتر MDM2 و تضعیف مسیر سرکوب گر تومور P53 و تشدید تشکیل تومور در انسان
پروتئین سرکوب گر تومور P53 طی تنشهای سلولی مانند آسیب DNA و فعال شدن انکوژن فعال میشود و برنامه رونویسی را که منجر به ترمیم DNA، توقف چرخه سلولی و در برخی موارد آپپتوز میشود، شروع میکند. مسیر پاسخ استرس P53 برای جلوگیری از تشکیل تومور بسیار ضروری است؛ بهعنوان مثال، هم در موش و هم در انسان که موتاسیون غیرفعال ژرملاین[31] را در یکی از آللهای ژن P53 حمل میکنند، تومور در سنین بسیار پایین زندگی و بهطور چشمگیری با فراوانی بالا گسترش پیدا میکند. موتاسیونهای غیرفعالکنندهی سوماتیک[32] در ژن p53 در بیش از 50 درصد تومورهای انسانی یافت میشوند. رویهمرفته، این مشاهدات و بسیاری از گزارشهای دیگر، اهمیت مسیر p53 را در سرکوب تومور حمایت میکند؛ بنابراین این منطقی است که فرض کنیم بهطور طبیعی واریانتهای ژنتیکی پلیمورفیک در گرههای اصلی مسیر p53 میبایستی زمینهی تغییر دیده شده در اشخاص، بین حساسیتشان به سرطان و پیشروی بیماریشان باشد. تحقیق برای تغییر ژنتیکی در مسیر p53 با نگاه به ژن MDM2 شروع شد که تنظیمکننده منفی مهم P53 را کد میکند. ژن MDM2 بهطور مستقیم متصل میشود و P53 را با تنظیم موقعیت، دوام و فعالیتش بهعنوان یک فعالکننده رونویسی مهار میکند. ژن MDM2 یک ژن ضروری در نمو موش مورین است بهطوریکه جنین قبل از قرار گرفتن در رحم میمیرد. این فنوتیپ کشنده با بلوکه کردن ژن P53 بهوجود نمیآید که مسلماً نشاندهندهی یک برهمکنش ژنتیکی مهم بین دو ژن در نمو موش مورین است. مندریسا[33] و همکاران (2003) اهمیت این برهمکنش را در موش بالغی که بهطور ژنتیکی تغییر یافته بود و سطح کاهشیافتهای از MDM2 را تولید میکرد، نشان دادند. این موشها کوچک، لنفوپنیک[34]، حساس به امواج رادیویی و دارای آپپتوز در سلولهای لنفوسیتی و اپیتلیال بودند. این فنوتیپها همگی نشاندهنده مستقل بودن P53 بودند، از این رو بیشتر نشاندهندهی این است که MDM2 یک تنظیمکننده کلیدی منفی P53 در موشهای در حال نمو و موشهای بالغ است. در انسان، مجموعهی تومورها mRNA و پروتئین MDM2 را بیش از حد بیان میکنند و این بیان بیش از حد با پیشروی سرطان تسریعیافته و فقدان پاسخ به درمان در ارتباط است. در زیرمجموعه این تومورها، بیان بیش از حد MDM2 متقابلاً منحصر به موتاسیون p53 بود که میتواند پیشنهاد کند بیان بیش از حد MDM2 میتواند جایگزینی برای p53 غیرفعالکننده توسط موتاسیون باشد. همانطور که بیان MDM2 بهنظر میرسد برای پاسخ p53 ضروری است، بهطور طبیعی تغییرات توالی که در پروموتر MDM2 رخ میدهد ممکن است منجر به تغییر بیان پروتئین MDM2 شود و از این رو سرکوبکننده تومور p53 و بهطور بالقوه سرطان در انسان را تحت تأثیر قرار میدهد (32).
SNP مربوط به ژن RAD51 خطر سرطان را در حاملین BRCA2 تغییر میدهد
موتاسیونهای ژرملاین در ژنهای BRCA1 و BRCA2 حساسیت به سرطان سینه و تخمدان را افزایش میدهند. نفوذ این موتاسیونها ناکامل بوده و وابسته به سن است، بنابراین خطر سرطان در حاملین با سن شروع به افزایش میکند، هرچند میانگین سن تشخیص سرطان در حاملین ژنهای BRCA1 و BRCA2 در مقایسه با غیرحاملین کمتر است. تخمین قابلیت نفوذ، شاید در نتیجه طرحهای اثباتی مختلف و یا اثرات آللی بهطور گسترده متغیر است. در خانوادهها مشخص شده افراد مبتلا به چندین مورد، برای تجزیه و تحلیل ارتباط مناسبند، خطر سرطان در طول عمر (تا سن 70 سالگی) حدود 85 درصد برای هر دو حاملین ژنهای BRCA1 و BRCA2 بود، 63 درصد برای سرطان تخمدان در حاملین BRCA1 بود و 27 درصد برای سرطان تخمدان در حاملین BRCA2 بود.
در مطالعات انجامشده در خانوادههای کمتر انتخابی و یا در سطح جمعیت، ریسک دائم 36-6 درصدی سرطان پستان و ریسک 16 درصدی ابتلا به سرطان تخمدان مشهود بود. این مطالعات در گروههای قومی مختلف انجام شد که تعداد محدودی از موتاسیونهای خاص را حمل میکردند و بنابراین میتوانستند نمایندهی این آللها باشند، در عوض نفوذ کلی موتاسیونهای BRCA1 و BRCA2 را منعکس میکنند. این تفاوتها پیشنهاد میکند که نفوذ موتاسیونهای BRCA1 و BRCA2 با فاکتورهای محیطی و ژنتیکی دیگر تغییر میکند. شناسایی چنین تغییردهندههایی پیامدهای مهمی مانند تسهیل ارزیابی دقیقتر ریسک در حاملانی که با انتخابهای بالینی دشوار در مورد ماستکتومی پیشگیرانه[35] و اووفورکتومی[36] مواجهند، دارد. ژن های مناسب جهت اصلاح شامل ژنهایی هستند که محصولات آنها در واکنش با BRCA1 و BRCA2 شرکت می کنند.
RAD51 هومولوگ RecA در باکتریهاست که برای میوز و نوترکیبی میوزی و ترمیم نوترکیبی شکستهای DNA دو رشتهای موردنیاز است. هردوی BRCA1 و BRCA2 نشان دادهاند که میتوانند با RAD51 واکنش نشان دهند و بلوکه کردن فنوتیپ BRCA1 و BRCA2 موشها مشابه بلوکه شدن RAD51 است. یک موتاسیون بیمعنی در RAD51 در دو بیمار ژاپنی با سرطان دوطرفه سینه توصیف شده است، وانگ و همکاران شفاهاً مدرکی ارائه دادند که یک SNP در ناحیه ترجمهنشونده 5Ꞌ ژن RAD51 با افزایش ریسک سرطان سینه در حاملین BRCA1 و BRCA2 در ارتباط است اما تأثیری در ریسک سرطان سینه در زنان فاقد BRCA1 و BRCA2 ندارد. این SNP بهصورت 135g/c طراحی شده است که جایگزینی G با C در موقعیت 135 cDNA ژن RAD51 انسانی است (33).
SNP در ژنهای انتقالی ABCC5 و ABCG1 با سمیتهای معدهای– رودهای مرتبط با ایرینوتیکن در بیماران مبتلا به سرطان کولورکتال ارتباط دارد
ایرینوتیکن[37] (با فرمول 7-ethyl-10-[4-(1-piperidino)]-1-piperidino) یک داروی مورد تأئید سازمان غذا و داروی آمریکا و آژانس دارویی اروپا برای درمان متاستاز بیماران دارای سرطان کولون[38] است. در حال حاضر، ایرینوتیکن یا بهتنهایی و یا در ترکیب با دیگر عوامل شیمیدرمانی مانند فلورواوراسیل[39]، بواسیزومب[40] و اگزالیپلاتین[41] در درمان خط اول یا دوم بیماران دارای سرطان کولون مورد استفاده قرار میگیرد. مصرف این دارو در سمیتهای شدید مانند نوتروپنیا و اسهال نوع تأخیری و حاد که نیاز به نظارت نزدیک و درمان فوری دارد، محدود میشود.
پروفایــــــــل سمیت ایرینوتیکن وابستــــــــــه به دوز دارو و برنامه است اما در تمام رژیمها اسهال شدید و نوتروپنی مهمترین سمیتهای محدودکننــــــــده دوز دارو هستند. ایــــــــــــرینوتیکن توسط سیتوکروم p450 CYP3A4 به
APC (7-ethyl-10-(4-N-[5-aminopentanoicacid]-1-piperidino)carbonyloxycamptothecin) و NPC (7-ethyl-10-(4-amino-1-piperidino) سمیتزدایی میشود و توسط کربوکسیل استرازهای CES1 و CES2 به متابولیتهای فعالش SN-38 تبدیل میشود، سپس با اسید گلوکورونیک به شــــــــکل SN-38G توسط آنــزیم UDP- گلوکورونوزیل ترانسفراز، UDP-گلوکورونوزیل ترانسفراز 1A1 و احتمالاً دیگر ایزوفرمها کانجوگه میشود که این آنزیمها همچنین گلوکورونیداسیون بیلیروبین را نیز کاتالیز میکنند. فعالیت ناقص گلوکورونیداسیون آنزیم UDP-گلوکورونوزیل ترانسفراز 1A1 با سطح سرمی بالای SN-38 و بیلیروبین در ارتباط است و منجر به سمیت میشود. اعضای خانواده ترانسپورتر کاست متصل به ATP، خروج محصولات متابولیک ایرینوتکان را تنظیم میکنند (شکل 2).
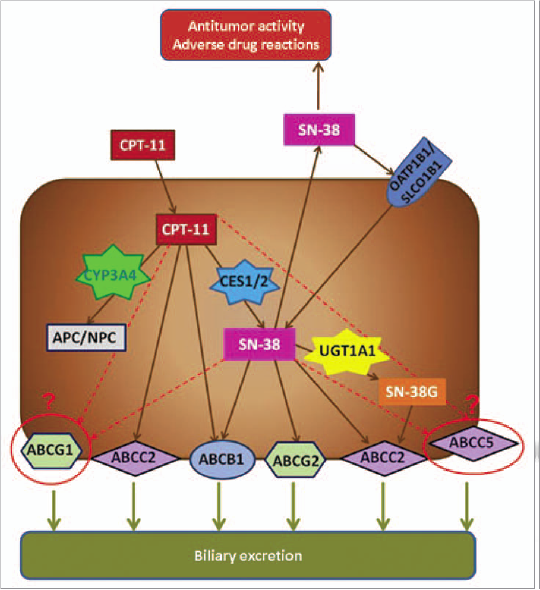
شکل 2: فعالیت ایرینوتکان و مسیر طبیعی
نتایج حاصل از مطالعهی مربوطه بهصورت دوایر گرد و نقطهچین نشان داده شده است
گزارش شده است که متغیرهای بین فردی در فارماکوکنتیک ایرینوتکان و SN-38 و همچنین پلیمورفیسم ژنتیکی آنزیم UGT1A1 در گلوکورونیداسیون SN-38 دخیلند. این قابلیت تغییر با تفاوتهای قابلتوجهی در نتیجه درمان و سمیت غیرقابل پیشبینی شدید در برخی از بیماران همراه است. بیماران هموزیگوت برای آلل 7/TA (UGT1A1 * 28) که دارای تکرار TA اضافی در منطقه پروموتر UGT1A1 هستند، بیشترین تغییرپذیری را در فارماکوکینیتیک ایرینوتکان و سمیتهای بالاتر، بخصوص نوتروپنی نشان میدهند. در سال 2005، این یافتهها منجر به اصلاح در برچسب داروی ایرینوتکان توسط FDA شد که شامل کاهش دوز در بیماران هوموزیگوت برای UGT1A1 * 28 بود. برای شناسایی ژنوتایپهای بیمار بهمنظور هدایت پزشکان به دوز مناسب ایرینوتکان، تست ژنتیک مورد تأئید قرار گرفت.
علاوه بر این، همانطور که مطالعات جمعیت، آللهای مختلف UGT1A1 را با سمیت با ایرینوتکان مرتبط میدانند و ایرینوژنومیک شامل چندین نوع مختلف ژنی است، رویکردهای نوآورانه بهشدت موردنیاز است تا دریچهی جدیدی را در این حوزهی فارماکوژنتیک داروی ضدسرطان و همچنین مصرف دارو باز کند. در مطالعهی دی مارتینو[42] و همکاران، با استفاده از یک پلتفرم میکروآرایه نوآورانه، پروفایل ایرینوژنومیک تمام ژنهای حاضر که در متابولیسم دارو درگیر بودند را شناسایی کردند و در یک سری از بیماران mCRC با سمیت رودهای- معدهای ناشی از ایرینوتکان را در مقایسه با کنترلهای همسان بدون تجربه سمیت رودهای- معدهای مقایسه نمودند. پلتفرم DMET Plus اجازه میدهد که تمام پليمورفيسمهاي شناختهشده در جذب، توزيع، متابوليسم و حذف آنزیمهای مرتبط با ADME روی يک آرایه تکميل شود (34).
منابع:
- Stenson, PD; Mort, M, Ball, EV, Howells, K, Phillips, AD, Thomas, NS, Cooper, DN (2009-01-22). “The Human Gene Mutation Database: 2008 update.”. Genome medicine 1(1): 13. PMID19348700.
- Ammitzbøll, Christian Gytz; Kjær, Troels Rønn; Steffensen, Rudi; Stengaard-Pedersen, Kristian; Nielsen, Hans Jørgen; Thiel, Steffen; Bøgsted, Martin; Jensenius, Jens Christian (28 November 2012). “Non-Synonymous Polymorphisms in the FCN1 Gene Determine Ligand-Binding Ability and Serum Levels of M-Ficolin”. PLoS ONE. 7 (11): e50585. PMC3509001 . PMID 23209787. doi:1371/journal.pone.0050585.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al (2001) Initial sequencing and analysis of the human genome. Nature 409: 860 – 921
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al (2001) The sequence of the human genome. Science 291: 1304 – 1351
- Risch NJ (2000) Searching for genetic determinants in the new millennium. Nature 405: 847 – 856
- Botstein D, Risch N (2003) Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat Genet 33(Suppl): 228 – 237
- Kruglyak L, Nickerson DA (2001) Variation is the spice of life. Nat Genet 27: 234 – 236 Krynetski EY, Tai HL, Yates CR, Fessing MY, Loennechen T, Schuetz JD, Relling MV, Evans WE (1996) Genetic polymorphism of thiopurine S-methyltransferase: clinical importance and molecular mechanisms. Pharmacogenetics 6: 279 – 290
- Carlson CS, Eberle MA, Rieder MJ, Smith JD, Kruglyak L, Nickerson DA (2003) Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans. Nat Genet 33: 518 – 521
- Bonnen PE, Wang PJ, Kimmel M, Chakraborty R, Nelson DL (2002) Haplotype and linkage disequilibrium architecture for human cancerassociated genes. Genome Res 12: 1846 – 1853
- Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22: 139 – 144
- Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV (2003) Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet 12: 205 – 216
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296: 2225 – 2229
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29: 229 – 232
- Stram DO, Leigh Pearce C, Bretsky P, Freedman M, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Thomas DC (2003) Modeling and E-M estimation of haplotype-specific relative risks from genotype data for a case-control study of unrelated individuals. Hum Hered 55: 179 – 190
- Yin J, Rockenbauer E, Hedayati M, Jacobsen NR, Vogel U, Grossman L, Bolund L, Nexo BA (2002) Multiple single nucleotide polymorphisms on human chromosome 19q13.2-3 associate with risk of Basal cell carcinoma. Cancer Epidemiol Biomarkers Prev 11: 1449 – 1453
- Choi EH, Foster CB, Taylor JG, Erichsen HC, Chen RA, Walsh TJ, Anttila VJ, Ruutu T, Palotie A, Chanock SJ (2003) Association between chronic disseminated candidiasis in adult acute leukemia and common IL4 promoter haplotypes. J Infect Dis 187: 1153 – 1156
- Colhoun HM, McKeigue PM, Davey Smith G (2003) Problems of reporting genetic associations with complex outcomes. Lancet 361: 865 – 872
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003) Metaanalysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33: 177 – 182
- Wacholder S, Rothman N, Caporaso N (2000) Population stratification in epidemiologic studies of common genetic variants and cancer: quantification of bias. J Natl Cancer Inst 92: 1151 – 1158
- Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A (2001) Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet 69: 831 – 843
- Cascorbi I, Henning S, Brockmoller J, Gephart J, Meisel C, Muller JM, Loddenkemper R, Roots I (2000) Substantially reduced risk of cancer of the aerodigestive tract in subjects with variant-463A of the myeloperoxidase gene. Cancer Res 60: 644 – 649
- Le Marchand L, Seifried A, Lum A, Wilkens LR (2000) Association of the myeloperoxidase 463G-a polymorphism with lung cancer risk. Cancer Epidemiol Biomarkers Prev 9: 181 – 184
- Golka K, Prior V, Blaszkewicz M, Bolt HM (2002) The enhanced bladder cancer susceptibility of NAT2 slow acetylators towards aromatic amines: a review considering ethnic differences. Toxicol Lett 128: 229 – 241
- Hein DW (2002) Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res 506 –507: 65 – 77
- Rebbeck TR, Jaffe JM, Walker AH, Wein AJ, Malkowicz SB (1998) Modification of clinical presentation of prostate tumors by a novel genetic variant in CYP3A4. J Natl Cancer Inst 90: 1225 – 1229
- Paris PL, Kupelian PA, Hall JM, Williams TL, Levin H, Klein EA, Casey G, Witte JS (1999) Association between a CYP3A4 genetic variant and clinical presentation in African-American prostate cancer patients. Cancer Epidemiol Biomarkers Prev 8: 901 – 905
- Nicoloso MS, Sun H, Spizzo R, Kim H, Wickramasinghe P, Shimizu M, Wojcik SE, Ferdin J, Kunej T, Xiao L, Manoukian S. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer research. 2010 Apr 1;70(7):2789-98.
- Weinberg, R. A. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res., 49: 3713–3721, 1989.
- Barsky, S. H., Siegal, G. P., Jannotta, F., and Liotta, L. A. Loss of basement membrane components by invasive tumors but not by their benign counterparts. Lab. Investig., 49: 140–147, 1983.
- Lukashew, M. E., and Werb, Z. ECM signaling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol., 8: 437–441, 1998.
- Coussens, L. M., and Werb, Z. Matrix metalloproteinases and the development of cancer. Chem. Biol., 3: 895–904, 1996
- Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, Onel K. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004 Nov 24;119(5):591-602.
- Levy-Lahad E, Lahad A, Eisenberg S, Dagan E, Paperna T, Kasinetz L, Catane R, Kaufman B, Beller U, Renbaum P, Gershoni-Baruch R. A single nucleotide polymorphism in the RAD51 gene modifies cancer risk in BRCA2 but not BRCA1 carriers. Proceedings of the National Academy of Sciences. 2001 Mar 13;98(6):3232-6.
- Di Martino MT, Arbitrio M, Leone E, Guzzi PH, Saveria Rotundo M, Ciliberto D, Tomaino V, Fabiani F, Talarico D, Sperlongano P, Doldo P. Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: a DMET microarray profiling study. Cancer biology & therapy. 2011 Nov 1;12(9):780-7.
Abstract
Single Nucleotide Polymorphisms (SNPs) are a difference in a nucleotide order than its similar position in the DNA sequence. SNP are the changes and variations of natural sequence with high-density occurring in the genomes. SNPs are considered as a major genetic source of phenotypic changes within a species and are considered to be a significant genetic marker. SNPs have many applications, such as molecular markers in genetic researches and modifications, as a molecular marker in genetic studies of diseases and genomics, in genetic mapping, selection of markers, and so on. SNPs have been studied over the years in the sequences of genes associated with the risk of cancers. In this study, we intend to briefly describe the role of SNP in the risk of developing a variety of cancers.
[1] Single Nucleotide Polymorphisms
[4] synonymous polymorphism
[5] replacement polymorphism
[6] missense
[7] nonsense
[8] premature stop codon
[10] triallelic SNP
[11] bottleneck events
[12] population private variants
[13] linkage study
[14] susceptibility or outcome
[15] Bayesian approaches
[16] xenobiotic
[17] Myeloperoxidase
[18] N-acetyltransferase 1
[19] Thiopurine
[20] methyltransferase
[21] germ-line
[22] protein-coding gene
[23] multiple low-penetrance alleles
[24] high-penetrance
[25] Milena
[26] Posttranscriptional gene
[27] surrounding microenvironment
[28] tumorigenesis
[29] matrix metalloproteinase
[30] extracellular membrane
[31] germline inactivating mutation
[32] Somatic inactivating mutations
[33] Mendrysa
[34] lymphopenic
[35] prophylactic mastectomy
[36] oophorectomy
[37] Irinotecan
[38] metastatic colorectal cancer
[39] fluorouracil
[40] bevacizumab
[41] oxaliplatin
[42] Di Martino
ژنومیکس و کاربرد آن در تشخیص بیماریها (2)
نشانگرهای مولکولی و تكنيكهاي نقشهبرداري ژنومي و انگشتنگاري DNA
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام