
سندرم کابوکی
Kabuki Syndrome

شاهین اسعدی (دانشجوی دکتری تخصصی ژنتیک پزشکی)
کلیاتی از سندرم کابوکی
سندرم کابوکی که تحت عنوان سندرم ترکیبی کابوکی یا سندرم نیکاوا-کوراکی نیز شناخته میشود، یک اختلال مادرزادی بسیار نادر اطفال با منشأ ژنتیکی مشکوک است که با اختلالات چند دستگاه مختلف از جمله ویژگیهای متمایز صورت، تأخیر رشد، درجه ناتوانی فکری، اختلالات اسکلتی و کوتاهی قامت همراه است. سندرم کابوکی همچنین میتواند طیف گستردهای از اختلالات را شامل شود و ارگانهای مختلف بدن را تحت تأثیر قرار دهد.

شکل 1: تصویر کودک مبتلا به سندرم کابوکی همراه با ویژگیهای متمایز در صورت
علائم و نشانههای سندرم کابوکی
برخی از ویژگیهای سندرم کابوکی در هنگام تولد (مادرزادی) آشکار میشوند. یافتههای سندرم کابوکی از فردی به فرد دیگر میتواند متفاوت باشد. همانطور که گفته شد، سندرم کابوکی میتواند بسیاری از سیستمهای مختلف بدن را تحت تأثیر قرار دهد. کودکان مبتلا به سندرم کابوکی، ظاهر صورت متمایز که شامل شکاف پلکی، بیرونزدگی پلک پایین، مُژه برجسته، اَبروهای کمانی، بینی گسترده با نوک مسطح یا پهن و بزرگ و گوشهای بدشکل را آشکار میکنند. ظاهر متمایز صورت به آهستگی پیشرفت میکند و ممکن است بیش از چندین سال طول بکشد. ویژگیهای اضافی صورت شامل یک اثر خفیف از رنگ مایل به آبی در کاسه سفیدی چشم (صلبیه آبی)، افتادگی پلک بالایی (پتوزیس)، چشم منحرف (استرابیسم)، سقف دهان بلند و قوسی شکل یا شکاف کام، فرورفتگی داخل لب پایین (چاله لب) و یک فک غیرعادی کوچک است که ممکن است در مبتلایان سندرم کابوکی رخ دهد.
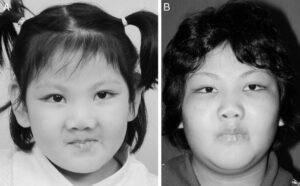
شکل 2: تصاویر مبتلایان سندرم کابوکی همراه با اختلالات مربوطه در صورت
کاهش رشد در مبتلایان سندرم کابوکی که معمولاً در سال اول زندگی آشکار میشود، شایع است. کاهش رشد با افزایش سن کودکان نیز بیشتر میشود و در نهایت منجر به کوتاهی قد و قامت در مبتلایان میشود. در موارد نادر، برخی از کودکان مبتلا ممکن است کمبود هورمون رشد نسبی داشته باشند. علاوه بر کاهش رشد، کودکان مبتلا به سندرم کابوکی ممکن است ناتوانی ذهنی را به فرم خفیف تا متوسط نشان دهند. ناتوانی فکری شدید در این سندرم بسیار نادر است و برخی کودکان نیز حتی بدون ناتوانی ذهنی هستند. برخی از کودکان مبتلا به سندرم کابوکی ممکن است تشنج، اندازه کوچک سر (میکروسفالی) و کاهش تون عضلانی (هایپوتونی) را تجربه کنند. تشنج میتواند بلافاصله بعد از تولد (دوران نوزادی) و یا در اواخر سن 12 سالگی، توسعه پیدا کند. برخی از کودکان مبتلا ممکن است تأخیر در سخن گفتن یا تکلم را تجربه کنند که اختلالات کام و از دست دادن حس شنوایی ممکن است به این تأخیر تکلم کمک کند. برخی دیگر از کودکان مبتلا به سندرم کابوکی ممکن است اختلالات رفتاری از جمله اضطراب و تمایل به تمرکز بر روی اشیاء را از خود نشان دهند. همچنین برخی از کودکان مبتلا به سندرم کابوکی ممکن است به موسیقی یا اَصوات خاصی علاقه داشته باشند و از برخی محرکها مانند برخی صداها یا بوها بیزار باشند. این در حالی است که علاقهمندی به موسیقی در مبتلایان سندرم کابوکی تقریباً، ژانر یا سَبکِ یکسانی را برای تمام نژادهای انسانی مبتلا به این سندرم نشان میدهد.

شکل 3: تصویر کودک مبتلا به سندرم کابوکی و نمای شماتیک از نحوه آرایش چشم بازیگران تئاتر نمایشی کابوکی (تئاتر انحصاری ژاپنیها) که گوشه چشم آنها شبیه گوشه چشم مبتلایان سندرم کابوکی است
علاوه بر این، کودکان مبتلا به سندرم کابوکی ممکن است مشکلات تغذیه از جمله ریفلاکس معده به مری، توانایی ضعیف در مکیدن شیر مادر از پستان و مشکل جذب یا هضم مواد مغذی از مواد غذایی (سوء جذب) را تجربه کنند، در نتیجه، بسیاری از کودکان مبتلا به این سندرم ممکن است افزایش وزن و رشد در نرخ مورد انتظار را نداشته باشند. برخی از کودکان مبتلا ممکن است در معرض عفونتهای مکرر تنفسی فوقانی و ذاتالریه و عفونتهای مکرر گوش میانی (اوتیت مدیا) که ممکن است به از دست دادن حس شنوایی کمک کند، قرار بگیرند.
اختلالات دندانی مانند دندانهای ریختهشده بهصورت خودبخودی، یا دندانهای بدشکل و یا منحرف نیز ممکن است در کودکان مبتلا به سندرم کابوکی رخ دهد، همچنین ناخن پا بهصورت کوچک یا نازک نیز گاهی اوقات در کودکان مبتلا به سندرم کابوکی دیده میشود.
اختلالات اسکلتی نیز ممکن است در مبتلایان سندرم کابوکی رخ دهد، از جمله انگشتان غیرطبیعی کوتاه پا، خمیدگی انگشتان، صافی کف پا، شُلشدگی مفاصل، اختلالات مهرههای کمر (دیسک کمر) و گردن، ناهنجاری جمجمه، اِنحنای غیرطبیعی ستون فقرات (اسکولیوز یا کیفوز) و جابجایی باسن و استخوان کشکک نیز ممکن است در مبتلایان سندرم کابوکی رخ دهد.
برخی از کودکان مبتلا به سندرم کابوکی ممکن است اختلالات خاص قلبی در بدو تولد (نقص مادرزادی قلب) را آشکار کنند. دو نقص قلبی در مبتلایان سندرم کابوکی شایع هستند که شامل تنگی شریان اصلی بدن (کوآرکتاسیون آئورت) و حفره در غشاء (سپتوم) که اتاق قلب (نقص بین دیواره دهلیزی و بطنی) را از هم جدا میکند، است.
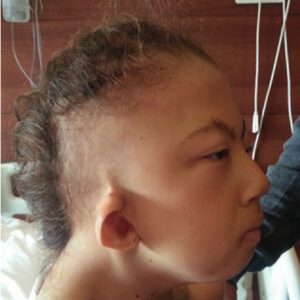
شکل 4: تصویر کودک مبتلا به سندرم کابوکی همراه با بدشکلی گوشها و اَبروهای کمانی
در برخی دیگر از مبتلایان سندرم کابوکی، ممکن است اختلالات کلیه از جمله ناهنجاری یا عدم توسعه کلیهها (دیسپلازی کلیوی و یا هایپوپلازی کلیوی)، اِنسداد جریان طبیعی اِدرار از کلیهها (هیدرونفروز) و تشکیل کلیه نعل اسبی رخ دهد. اختلالات دستگاه گوارش شامل پیچش نامناسب یا ناهنجار روده بزرگ و عدم وجود یا اِنسداد مقعد (آنوس) نیز ممکن است در مبتلایان سندرم کابوکی رخ دهد.
کاهش عملکرد مناسب سیستم ایمنی نیز در مبتلایان سندرم کابوکی گزارش شده است. زنانی که تحت تأثیر این سندرم هستند ممکن است رشد زودرس پستان را تجربه کنند، این در حالی است که برخی از مردان مبتلا به سندرم کابوکی ممکن است بیضه نزولنکرده یا توسعهنیافته را تجربه کنند. برخی دیگر از مبتلایان سندرم کابوکی ممکن است افزایش وزن و بلوغ سریع داشته باشند.

شکل 5: نمای شماتیک از گریم و آرایش بازیگران تئاتر نمایشی کابوکی در ژاپن که شباهت زیادی با چهره مبتلایان سندرم کابوکی دارد
علتشناسی سندرم کابوکی
در ماه اوت سال 2010، گروهی از محققان در دانشگاه واشنگتن گزارش کردند که جهش در ژن KMT2D (نام قبلی MLL2)، مسئول سندرم کابوکی در اکثر مبتلایان این اختلال ژنتیکی است.
در سال 2012 گروهی از محققان کشور بلژیک، ژن دومی را با نام KDM6A برای ایجاد سندرم کابوکی شناسایی کردند. اغلب موارد سندرم کابوکی، در اثر جهش ژن KMT2D رخ میدهد که بدون سابقه خانوادگی به دلیل جهش جدید، ایجاد میشود. با این حال، وقوع سندرم کابوکی با سابقه خانوادگی نیز گزارش شده است. ژن KMT2D در بازوی بلند کروموزوم شماره 12 بهصورت 12q13.12 مستقر است. ژن KDM6A در بازوی کوتاه کروموزوم جنسی X بهصورت Xp11.3 مستقر است. سندرم کابوکی که در اغلب موارد توسط جهش ژن KMT2D ایجاد میشود، از الگوی توارثی اتوزومال غالب پیروی میکند. این در حالی است که اگر فردی با جهش در ژن KDM6A دچار سندرم کابوکی گردد، از الگوی توارثی وابسته به X غالب پیروی خواهد کرد؛ بنابراین شانس داشتن فرزند مبتلا به سندرم کابوکی توسط دو الگوی اتوزومال غالب و وابسته به X غالب، به میزان 50% برای هر بارداری احتمالی خواهد بود.

شکل 6: نمای شماتیک از کروموزوم شماره 12 که ژن KMT2D در بازوی بلند این کروموزوم بهصورت 12q13.12 مستقر است
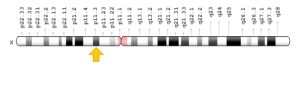
شکل 7: نمای شماتیک از کروموزوم جنسی X که ژن KDM6A در بازوی کوتاه این کروموزوم بهصورت Xp11.3 مستقر است
فراوانی سندرم کابوکی
سندرم کابوکی، مردان و زنان را به تعداد مساوی تحت تأثیر قرار میدهد. بروز سندرم کابوکی ناشناخته است اما تخمین زده میشود که فرکانس شیوع این سندرم حدود 1 در 32000 یا 1 در 86000 تولد زنده میان جمعیت عمومی مردم باشد. تا به امروز بیش از 300 مورد از مبتلایان سندرم کابوکی در ادبیات پزشکی گزارش شده است. اگرچه این اختلال اولین بار در ژاپن گزارش شده است، اما از آن زمان به بعد سندرم کابوکی در میان نژادهای مختلف انسانی نیز گزارش گردیده است.
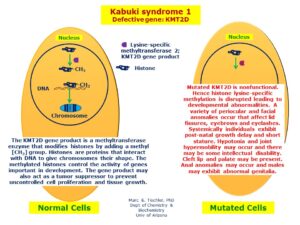
شکل 8: نمای شماتیک از مکانیسم بیان و اثرگذاری ژن KMT2D در سلولهای نرمال و جهشیافته مبتلا به سندرم کابوکی
تشخیص سندرم کابوکی
سندرم کابوکی بر اساس یافتههای بالینی و فیزیکی مبتلایان، از جمله ویژگیهای متمایز صورت، اختلالات اسکلتی، ناتوانی ذهنی، اختلالات انگشتان و کوتاهی قد تا حدودی قابل تشخیص است، اما تنها راه تشخیص سندرم کابوکی، آزمایش ژنتیک مولکولی برای بررسی وجود جهش یا عدم وجود جهش در ژنهای KMT2D و KDM6A است.
مسیرهای درمانی سندرم کابوکی
هیچ درمان خاص و مؤثری برای سندرم کابوکی وجود ندارد. مسیر درمانی این سندرم بر اساس علائمی که هر فرد مبتلا آشکار میکند، مشخص میشود. درمان ممکن است با تلاش و هماهنگی یک تیم از متخصصان شامل متخصص اطفال، جراحان، متخصص دندان، آسیب شناسان گفتاری، متخصص شنواییسنجی، متخصص چشم، متخصص تغذیه، متخصص گوارش و هورمون، متخصص ژنتیک مولکولی یا ژنتیک پزشکی و دیگر متخصصان مراقبتهای بهداشتی انجام پذیرد. مداخله زودهنگام برای بهبود وضعیت مبتلایان سندرم کابوکی بسیار حائز اهمیت است. مشاوره ژنتیک برای والدینی که سابقه خانوادگی به سندرم کابوکی یا هر نوع آسیب ژنتیکی و متابولیکی دارند و همچنین برای خانوادههایی که طالب فرزندی سالم و طبیعی هستند، امری ضروری است.
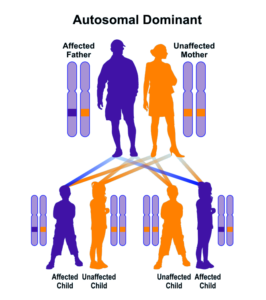
شکل 9: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم کابوکی با جهش ژن KMT2D از این الگو تبعیت میکند

شکل 10: نمای شماتیک از الگوی توارثی وابسته به X غالب تحت تأثیر کروموزوم جنسی X پدری که سندرم کابوکی در اثر جهش ژن KDM6A از این الگو پیروی میکند

شکل 11:نمای شماتیک از الگوی توارثی وابسته به X غالب تحت تأثیر کروموزوم جنسی X مادری که سندرم کابوکی در اثر جهش ژن KDM6A از این الگو نیز تبعیت میکند
تاریخچه سندرم کابوکی
سندرم کابوکی اولین بار در سال 1981 توسط دو گروه از محققان ژاپنی به رهبری نوریو نیکاوا و یوشیکازو کاروکی گزارش گردید. این اختلال در ابتدا به نام سندرم آرایش کابوکی معرفی گردید. دلیل نامگذاری آرایش کابوکی این بود که ویژگیهای صورت بسیاری از کودکان مبتلا به این سندرم، شبیه آرایش یا گریم بازیگران در شهر کابوکی ژاپن است که شکلی از تئاتر ژاپنی است. اصطلاح آرایش بعد از مدتی از این سندرم حذف شد و تنها نام منطقه کابوکی باقی ماند و تا به امروز، این اختلال را با نام سندرم کابوکی معرفی میکنند.


شکل 12: تصاویر کاشفان ژاپنی سندرم کابوکی در سال 1981
منبع:
اسعدی شاهین، جمالی مهسا، باقری رعنا، سادهدل سمانه، توحیدیراد مانوش، کتاب پاتولوژی در ژنتیک پزشکی 1 (A-L)، صفحات 653-645، انتشارات کتب دانشگاهی عمیدی، بهار 1396.
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام