
سندرم سیمسون- گلابی- بِهمِل
Simpson-Golabi-Behmel Syndrome
شاهین اسعدی (کارشناس ارشد ژنتیک)
کلیاتی از سندرم سیمسون- گلابی- بِهمِل
سندرم سیمسون- گلابی- بِهمِل، یک اختلال ژنتیکی است که بر بسیاری از قسمتهای مختلف بدن تأثیر میگذارد و عمدتاً در مردان اتفاق میافتد. این سندرم بهعنوان سندرم رشد بیش از حد نیز شناخته میشود، چرا که نوزادان آسیبدیده در هنگام تولد بزرگتر از حالت طبیعی (ماکروزومی) هستند و رشد بیش از حد دارند و میزان وزن خود را با نرخ غیرمعمول افزایش میدهند.
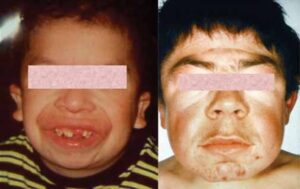
شکل 1: تصاویر کودکان مبتلا به سندرم سیمسون- گلابی- بِهمِل همراه با ویژگیهای مشخص صورت
علائم و نشانههای بالینی سندرم سیمسون- گلابی- بِهمِل
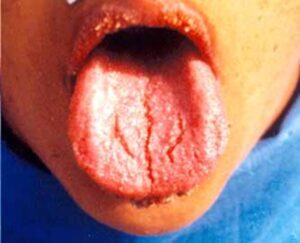
علائم و نشانههای این سندرم بهطور گستردهای متفاوت است؛ مبتلایان سندرم سیمسون- گلابی- بِهمِل با موارد خفیف، اغلب تا بزرگسالی نیز زندگی میکنند. افراد مبتلا دارای ویژگیهای مشخصی هستند از جمله فاصله چشمها بهطور گسترده از یکدیگر (هایپرتلوریسم)، دهان بیش از حد بزرگ (ماکروستومی)، زبان بیش از حد بزرگ (ماکروگلوسی)، بینی پهن با پیشانی گسترده، ناهنجاریهای سقف دهان (کام) و چهره بزرگ.
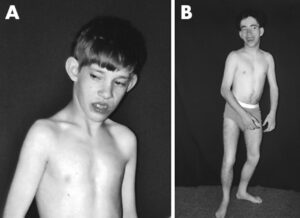
شکل 2: تصویر مبتلایان سندرم سیمسون- گلابی- بِهمِل همراه با اختلالات اسکلتی
سایر ویژگیهای سندرم سیمسون-گلابی-بِهمِل شامل اختلال قفسه سینه و شکم است. نوزادان آسیبدیده ممکن است با یک یا چند نوک پستان اضافی متولد شوند، باز شدن غیرطبیعی عضله شکم (دیستازیس رِکتی)، فتق بیضه و سوراخ در دیافراگم نیز از علائم دیگر این سندرم است.
علاوه بر این، افراد مبتلا ممکن است نقایص قلبی، کلیههای بزرگ غیرطبیعی، کبد و طحال بزرگ غیرطبیعی (هپاتوسپیلنومگالی) و اختلالات اسکلتی را تجربه کنند. همچنین این سندرم میتواند بر توسعه دستگاه گوارش، سیستم اِدراری و ژنتیال تأثیر بگذارد. برخی از مبتلایان این سندرم، ناتوانی فکری خفیف تا شدید را نیز تجربه میکنند، درحالیکه سایر افراد مبتلا به این سندرم دارای هوش طبیعی هستند.
شایان ذکر است که حدود 10 درصد از افراد مبتلا به سندرم سیمسون- گلابی- بِهمِل در دوران کودکی به تومورهای سرطانی یا غیرسرطانی مبتلا میشوند. شایعترین تومورها یک نوع نادر از سرطان کلیه است که به نام تومور ویلمز شناخته میشود و نوعی تومور دیگر به نام تومور نوروبلاستوما هم هست که از رشد بیش از حد سلولهای عصبی حاصل میگردد .


شکل 3: نمای کلی از علائم و نشانههای سندرم سیمسون- گلابی- بِهمِل
علتشناسی سندرم سیمسون- گلابی- بِهمِل
سندرم سیمسون- گلابی- بِهمِل در اثر جهش ژن GPC3 که در بازوی بلند کروموزوم جنسی X بهصورت Xq26.2 مستقر است، ایجاد میشود. این ژن دستورالعمل لازم برای سنتز پروتئینی به نام گلیپیکان 3 را فراهم میکند که مهارکننده مسیر رشدی به نام مسیر سیگنالینگ خارپُشت است. این مسیر، برای رشد و تقسیم سلولی (تکثیر)، تخصص سلولی و شکلگیری طبیعی الگوی بسیاری از قسمتهای بدن در طول رشد جنین حیاتی است. محققان معتقدند که پروتئین گلیپیکان 3 به ایجاد شکل بدن کمک میکند تا برخی از سلولها زمانی که دیگر موردنیاز نیستند، خود را از بین ببرند (آپوپتوزیس).

شکل 4: نمای شماتیک از کروموزوم جنسی X که ژن GPC3 در بازوی بلند این کروموزوم بهصورت Xq26.2 مستقر است

شکل 5: نمای شماتیک از کروموزوم جنسی X که ژن CXORF5 در بازوی کوتاه این کروموزوم بهصورت Xp22.2 مستقر است
جهشهای ژنی GPC3 مانع از تولید پروتئین گلیپیکان 3 میشود که همین امر نیز مانع مهار مسیر سیگنالینگ خارپُشتی میگردد؛ بنابراین بیشفعالی از این مسیر منجر به افزایش میزان رشد و تقسیم سلولی قبل از تولد میشود. این افزایش تکثیر سلولی حتی بهطور جزئی منجر به رشد بیش از حد بدن انسان در سندرم سیمسون- گلابی- بِهمِل میگردد. هنوز مشخص نیست که چگونه تغییرات در سیگنالینگ خارپُشتی موجب اختلالات دیگر در این سندرم میشود.
محققان اختلالی را شناسایی کردهاند که با ویژگیهای سندرم سیمسون- گلابی- بِهمِل همپوشانی دارد و بهعنوان سندرم سیمسون- گلابی- بِهمِل نوع 2 (SGBS2) شناخته میشود. سندرم سیمسون- گلابی- بِهمِل نوع 2 در اثر جهش ژن CXORF5 که در بازوی کوتاه کروموزوم جنسی X بهصورت Xp22.2 مستقر است، ایجاد میشود. علائم و نشانههای سندرم سیمسون- گلابی- بِهمِل نوع 2 شدیدتر از مواردی است که در سندرم سیسمون- گلابی- بِهمِل نوع 1 رخ میدهد و افراد مبتلا به این اختلال معمولاً در همان اوایل بعد از تولد جان خود را از دست میدهند.
شکل 6: نمای شماتیک از مسیر مولکولی ژن GPC3
سندرم سیمسون- گلابی- بِهمِل از الگوی توارثی وابسته به ایکس مغلوب پیروی میکند. زنان چون دارای دو کروموزوم جنسی ایکس هستند، بنابراین نسبت به بیماریهای وابسته به ایکس مغلوب بهصورت حامل میباشند و مردان چون دارای یک کروموزوم جنسی ایکس هستند، بنابراین با همان تکنسخه ژن جهشیافته موجود در کروموزوم جنسی ایکس، بیماری را بروز میدهند. شایان ذکر است که مردان مبتلا به بیماریهای وابسته به ایکس مغلوب هرگز آلل بیماری را به فرزندان پسر خود منتقل نمیکنند.
فراوانی سندرم سیمسون- گلابی- بِهمِل
فرکانس شیوع سندرم سیمسون- گلابی- بِهمِل ناشناخته است. تاکنون حدود 250 مورد مبتلا به این سندرم از سراسر جهان در ادبیات پزشکی گزارش شده است.
تشخیص سندرم سیمسون- گلابی- بِهمِل
سندرم سیمسون- گلابی- بِهمِل بر اساس یافتههای بالینی و فیزیکی مبتلایان و برخی آزمونهای آسیبشناسی تشخیص داده میشود. استفاده از تکنیکهای تصویربرداری رادیولوژیکی مانند سونوگرافی و سی.تی.اسکن میتواند در تشخیص سندرم مؤثر باشد. دقیقترین روش تشخیص سندرم بِهمِل، آزمایش ژنتیک مولکولی برای ژنهای GPC3 و CXORF5 بهمنظور بررسی وجود جهشهای احتمالی است. تشخیص پیش از تولد نیز با استفاده از مایع آمنیوسنتز و نمونهبرداری از پرزهای کوریونی جفت جنین در هفتههای تکوین بارداری امکانپذیر است.

شکل 7: نمای شماتیک از الگوی توارثی وابسته به X مغلوب (مادر ناقل) که سندرم سیمسون- گلابی- بِهمِل از این الگو تبعیت میکند
مسیرهای درمانی سندرم سیمسون- گلابی- بِهمِل
استراتژی درمان و مدیریت سندرم سیمسون- گلابی- بِهمِل بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص ارتوپدی، جراح، متخصص گوارش، متخصص مغز و اعصاب و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. هیچ درمان قاطعی برای سندرم سیمسون- گلابی- بِهمِل وجود ندارد و هرگونه اقدامات بالینی بهمنظور تخفیف رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند از جایگاه ویژهای برخوردار است.
تاریخچه سندرم سیمسون- گلابی- بِهمِل
سندرم سیمسون- گلابی- بِهمِل برای اولین بار توسط مهین گلابی، سیمسون و بِهمِل در سال 2006 گزارش گردید.

شکل 8: نمای شماتیک از الگوی توارثی وابسته به X مغلوب (پدر بیمار) که سندرم سیمسون- گلابی- بِهمِل نیز از این الگو پیروی میکند
منبع:
اسعدی شاهین، دکتر دادشپور مهدی، دکتر علیپور شهریار، جمالی مهسا، پورجعفر رؤیا، وحدانی کیا ویدا، فیروزی اکرم، باقری رعنا، سادهدل سمانه، قلیزاده زهرا، کتاب پاتولوژی در ژنتیک پزشکی 4 (M-W)، صفحات 921-915، انتشارات کتب دانشگاهی عمیدی، زمستان 96.
سندرم هایپوپلازی مادرزادی آدرنال وابسته به ایکس
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام