سندرم بیرت-هاگ-دوب
Birt–Hogg–Dubé Syndrome


شاهین اسعدی (دانشجوی ژنتیک مولکولی)، مهسا جمالی (کارشناس ارشد ژنتیک)
نگارنده مسئول: شاهین اسعدی (Molecular Geneticist)
کلیات
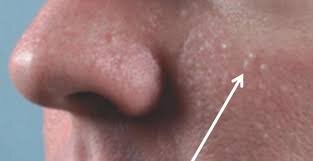
یک اختلال نادر ژنتیکی پوست است که با پاپول در پوست سر، صورت و نیمتنه بالایی همراه است. این تومورهای خوشخیم (هامارتوم) فولیکول مو، فیبروفولیکلوما نامیده میشوند. همچنین، سندرم بیرت-هاگ-دوب با توسعه کیستهای خوشخیم ریوی و افزایش بروز خطر سرطان کلیه همراه است.
شکل 1: تصویر مرد مبتلا به سندرم بیرت-هاگ-دوب، همراه با ضایعات پوستی پاپول در صورت
علائم و نشانهها
علائم و نشانههای سندرم بیرت-هاگ-دوب از فردی به فرد دیگر متفاوت است؛ شایعترین علائم عبارتند از: ضایعات متعدد خوشخیم پوست، کیست ریوی بهصورت پنوموتوراکس و تومورهای کلیوی (تومورهای بدخیم و خوشخیم). ضایعات پوستی شایعترین علائمی است که در حدود 85 درصد از مبتلایان سندرم بیرت-هاگ-دوب دیده میشود، بااینحال، در برخی از افراد مبتلا به این سندرم ممکن است کیست ریوی یا پنوموتوراکس و نئوپلازی کلیوی بدون ضایعات پوستی ایجاد شود.
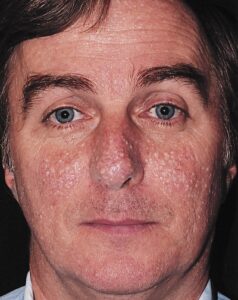
شکل 2: تصویر فرد مبتلا به سندرم بیرت-هاگ-دوب همراه با فیبروفولیکلوما در پوست صورت
ضایعات پوستی در سندرم بیرت-هاگ-دوب، بهعنوان فیبروفولیکلوما شناخته میشود و معمولاً بر روی پوست سر، صورت و گردن رخ میدهد، اما میتواند در نرمه گوش و مخاط دهان نیز یافت شود و شروع این ضایعات معمولاً بعد از سن 20 سالگی میباشد. شایان ذکر است که تعداد ضایعات پوستی بسته به سن افراد مبتلا نیز متفاوت خواهد بود و معمولاً با افزایش سن، تعداد این ضایعات پوستی هم افزایش مییابد، بطوریکه ممکن است در برخی از مبتلایان، تنها بهصورت یک یا چند زخم در پوست پدیدار شود، درحالیکه دیگران ممکن است صدها زخم از این ضایعات را در پوست خود داشته باشند.
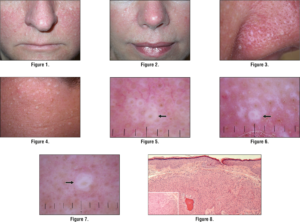
شکل 3: تصاویر ماکروسکوپی و میکروسکوپیک از ضایعات پوستی (پاپول) در صورت و پیشانی مبتلایان سندرم بیرت-هاگ-دوب
افراد مبتلا به سندرم بیرت-هاگ-دوب، ممکن است کیستهای متعدد ریوی را در یکی از ریهها یا هر دو ریه داشته باشند که در بیش از 80 درصد مبتلایان سندرم بیرت-هاگ-دوب رخ میدهد. این کیستها معمولاً بدون علائم هستند و عملکرد ریهها نیز طبیعی است، اما در حدود یکسوم از مبتلایان سندرم بیرت-هاگ-دوب، ممکن است کیستهای ریوی باعث اختلال در عملکرد ریهها بهصورت پنوموتوراکس گردد که در مواقعی میتواند تهدیدکننده حیات برای این مبتلایان باشد.
پنوموتوراکس یا هوا جنبی، به معنی وجود هوا در حفره جنب میباشد که مانع باز شدن کامل ریه میشود.
پنوموتوراکس دارای انواع مختلفی است که شامل پنوموتوراکس اولیه (خود بخودی) و ثانویه (باز) میباشد.
پنوموتوراکس اولیه: هوا از یک سوراخ یا شکافی در ساختمان داخلی دستگاه تنفس (مانند برونش، برونشیول، آلوئول) به درون فضای پلور راه مییابد. پاره شدن کیسههای هوایی کوچک در ریه در اثر آسم، آبسه، آمپیم ریه، آمفیزم گاهی علت مشخصی ندارد. شکسته شدن دندهها بهطور شایع منجر به هواجنبی بسته میشود.
پنوموتوراکس ثانویه: در پنوموتوراکس باز یا ثانویه هوا از طریق یک سوراخ در دیواره قفسه سینه یا دیافراگم وارد فضای پلور میگردد. پنوموتوراکس شدید معمولاً به دنبال ترومای قفسه صدری روی میدهد. زخمهای نافذ قفسه سینه مثل زخم چاقو یا گلوله و … اجازه ورود هوای آزاد به فضای جنب را داده و از این طریق باعث روی هم افتادگی ریه میشوند. بروز پنوموتوراکس در پنومونی، سل، عارضه کشیدن مایع از ریه (توراسنتز) و تهویه مکانیکی (PEEP) نیز ممکن است. وقتیکه هوا وارد حفره پلور شود و نتواند خارج گردد، پنوموتوراکس فشارنده پدید میآید و فشار در حفره پلور بالا میرود و باعث جابجایی مدیاستن به سمت مخالف طرف مبتلا میگردد. پنوموتوراکس در سندرم بیرت-هاگ-دوب، اغلب در افراد جوان و بهطور متوسط در سنین 7 تا 16 سال رخ میدهد.
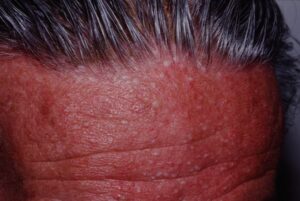
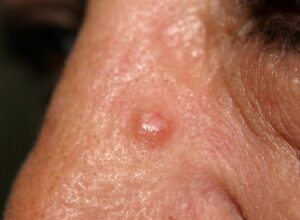
شکل 4: تصویر مرد مبتلا به سندرم بیرت-هاگ-دوب همراه با پاپولهای ظاهرشده در پوست پیشانی
حدود 15 تا 30 درصد از افراد مبتلا به سندرم بیرت-هاگ-دوب، ممکن است نئوپلاسمهای متعدد کلیوی را ایجاد کنند که این نئوپلاسمها معمولاً رشد آهسته دارند و هر دو کلیه را تحت تأثیر قرار میدهند. شایان ذکر است که متوسط سن تشخیص برای نئوپلازی کلیوی 48 تا 50 سال میباشد.
یافتههای اضافی دیگر در سندرم بیرت-هاگ-دوب شامل ضایعات در دهان (پاپول دهان)، تومورهای خوشخیم متشکل از بافت چربی (لیپوما)، تومورهای خوشخیم متشکل از بافت چربی که با رگهای خونی بزرگ غیرطبیعی (آنژیولیپوما) همراه است، تومور خوشخیم از غدد پاراتیروئید (آدنوما پاراتیروئید)، تومور خوشخیم غدد بزاقی (انکوسیتوما پاروتید) و یک خال مادرزادی متشکل از بافت همبند میباشد.
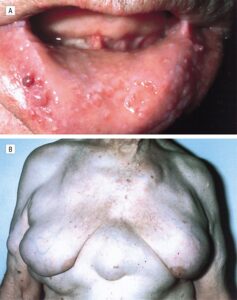
شکل 5: تصویر زن مبتلا به سندرم بیرت-هاگ-دوب همراه با پاپول در مخاط دهان و تومورهای لیپوما و آنژیولیپوما
علتشناسی
سندرم بیرت-هاگ-دوب در اثر جهش ژن FLCN که در بازوی کوتاه کروموزوم شماره 17 بهصورت 17p11.2 مستقر است، ایجاد میشود. ژن FLCN حاوی دستورالعمل سنتز پروتئین فولیکولین است که احتمالاً در مسیرهای سلولی منجمله رشد سلولی، تولید انرژی، سوختوساز بدن و تعامل سلولی نقش دارد. علاوه بر این، FLCN یک ژن سرکوبگر تومور است که میتواند رشد آهسته سلول را تنظیم کند و زمانی که سلولها دچار آسیب ژنتیکی شدهاند، میتواند در تعمیر DNA نقش داشته باشد و یا اینکه سلولهای آسیبدیده را به سمت مرگ فیزیولوژیک سلولی (Apoptosis) هدایت کند.
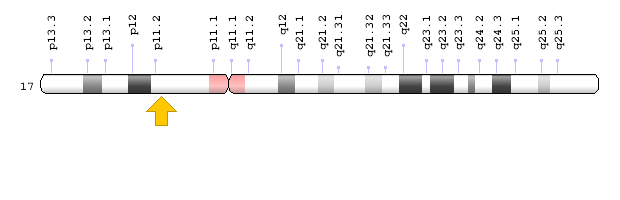
شکل 6: نمای شماتیک از کروموزوم شماره 17 که ژن FLCN در بازوی کوتاه این کروموزوم بهصورت 17p11.2 مستقر است
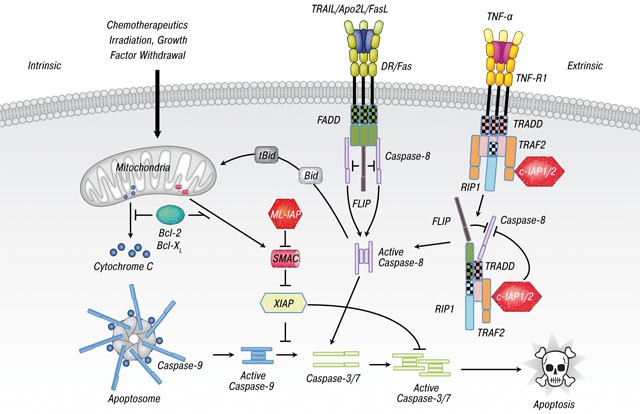
شکل 7: مسیر مولکولی مرگ فیزیولوژیک سلولی (Apoptosis)
هرگونه جهش در این ژن یا هر ژن سرکوبگر تومور، مستعد هدایت سلولها به سمت سرطان خواهد شد. سندرم بیرت-هاگ-دوب از الگوی توارثی اتوزومال غالب پیروی میکند، بنابراین برای ایجاد این سندرم تنها یک نسخه از ژن جهشیافته FLCN (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزند مبتلا به سندرم بیرت-هاگ-دوب برای هر بارداری احتمالی، به میزان 50% میباشد. لازم به ذکر است که در برخی موارد نیز، سندرم بیرت-هاگ-دوب در اثر تغییرات ژنتیکی خودبهخودی (جهش جدید) یا پراکنده ایجاد میشود.
فراوانی
یک اختلال ژنتیکی نادر است که مردان و زنان را به تعداد مساوی تحت تأثیر قرار میدهد. تاکنون کمتر از 300 خانواده مبتلا به سندرم بیرت-هاگ-دوب در ادبیات پزشکی گزارش شده است. ازآنجاکه اکثر موارد سندرم بیرت-هاگ-دوب به اشتباه تشخیص داده میشود، بنابراین تعیین دقیق فرکانس شیوع این سندرم در جهان دشوار است.
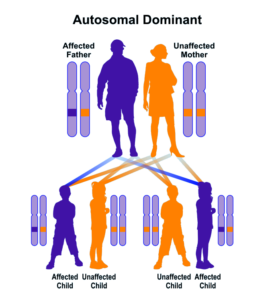
شکل 8: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم بیرت-هاگ-دوب از این الگو تبعیت میکند
تشخیص
براساس علائم و یافتههای مشخصه مانند پاپول دهان، فیبروفولیکوما، سابقه پنوموتوراکس خودبخودی و بپوسی بافتهای تومور کلیوی تشخیص داده میشود، همچنین استفاده از تکنیکهای تصویربرداری رادیویی مانند توموگرافی کامپیوتری (CT) یا رزونانس مغناطیسی (ام.آر.آی) میتوانند در تشخیص سندرم بیرت-هاگ-دوب کمککننده باشند. قطعیترین روش تشخیص سندرم بیرت-هاگ-دوب، آزمایش ژنتیک مولکولی برای ژن FLCN بهمنظور بررسی وجود جهشهای احتمالی میباشد.
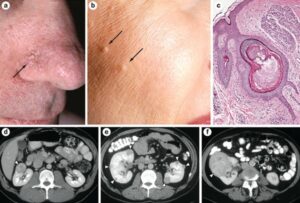
شکل 9: تصاویر رادیولوژی و میکروسکوپیک از حضور پاپول و فیبروفولیکلوما در پوست صورت مبتلایان
مسیرهای درمانی
درمان و مدیریت سندرم براساس علائم و نشانههایی است که هر فرد مبتلا، آن را آشکار میکند. درمان ممکن است شامل لیزر برای از بین بردن بافت پوست آسیبدیده، باشد که معمولاً ضایعات پوستی را با موفقیت از بین میبرد، اما اغلب، ضایعات دوباره ظهور میکنند. عمل جراحی برای بهبود وضعیت ریهها (آسپیراسیون) و عمل جراحی برای حذف نئوپلاسمهای کلیوی (نفرکتومی) نیز میتواند در بهبود وضعیت سلامتی مبتلایان سندرم بیرت-هاگ-دوب مؤثر باشد. مشاوره ژنتیک نیز برای تمامی والدینی که خواستار فرزندی سالم و نرمال هستند، از جایگاه ویژهای برخوردار است.
تاریخچه
اولین بار در سال 1977 توسط سه پزشک کشور کانادا به نامهای دکتر بیرت، دکتر هاگ و دکتر دوب در ادبیات پزشکی گزارش گردید.

شکل 10: تصویر دکتر آرتور بیرت پزشک اهل کشور کانادا، یکی از کاشفان سندرم بیرت-هاگ-دوب
منابع:
اسعدی شاهین، جمالی مهسا، باقری رعنا، سادهدل سمانه، توحیدی راد مانوش، کتاب پاتولوژی در ژنتیک پزشکی جلد اول (A-L)، صفحات 174-165، انتشارات کتب دانشگاهی عمیدی، بهار 1396
https://www.cancer.net/cancer-types/birt-hogg-dube-syndrome
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام