
آزمایشگاه و بالین
سرطانهای فامیلی و توارثی
دکتر محسن منشدی
آزمایشگاه تشخیص طبی دکتر منشدی

بهنظر میرسد اکثر سرطانها بهعلت تغییراتی که در طول عمر فرد مبتلا رخ میدهد، بوجود میآیند. برخی از سرطانها وابسته به ژنهای معیوب (جهشیافته) هستند که از والدین فرد به ارث می رسند. در واقع، این سرطان نیست که به ارث میرسد، بلکه ژنهای معیوب هستند که باعث میشوند احتمال ابتلا به سرطانهای خاص بیشتر شود. خانوادههایی که دارای این ژنها هستند احتمال ابتلا به سرطان در آنها بسیار بیشتراست.
فهرست جهشهای شایع ژنتیکی در سرطانهای فامیلی و توارثی از مؤسسه ملی سرطان، شامل جهشهایی است که بر چندین ژن سرکوبکننده تومورهای مختلف تأثیر میگذارند. سرطانهای ناشی از تغییرات ارثی در ژنهای سرکوبکننده تومور شامل سرطان پستان، سرطان تخمدان، سرطان پروستات، لوسمی، سرطان پانکراس و سرطان روده بزرگ است (1) (2) (3).
نکته: فقط قسمتی از هریک از این سرطانها با جهشهای ارثی مرتبط است بقیه موارد اسپورادیک بوده و ارثی نیستند.
درحال حاضر بسیاری از سرطانها برای بررسی درمان با داروهای موردنظر بهطور مرتب تحت آزمایش قرار میگیرند، این دسته شامل برخی از موارد مرتبط با سندرمهای سرطان ارثی و فهرست رو به رشدی از سرطانهای دیگر است.
از آنجا که ابتلا به سرطانهای نامبرده در لیست زیر، میتواند بهعلت جهشهای ژنی ارثی باشد، بسیاری از افراد مبتلا به آنها برای بررسی وجود ژن موردنظر، انتخاب میشوند (4) (5).
- سرطان ارثی پستان
- سندرم لی– فرامینی
- سندرم کاودن
- سندرم لینچ
- پولیپوز آدنوماتوز فامیلی
- رتینوبلاستوم
- نئوپلازی اندوکرین چندگانه
- سندرم فون هیپل– لیندو
در زیر شرح مختصری از موارد بهتر شناختهشده سرطانهای ارثی داده خواهد شد:
سرطان پستان و تخمدان ارثی
ژنهای: BRCA1 ,BRCA2
انواع سرطانهای مرتبط: سرطانهای پستان و تخمدان در خانمها و سرطانهای دیگری از قبیل سرطانهای پروستات، پانکراس و سرطان پستان در مردان
BRCA1 و BRCA2 ژنهای سرکوبگر تومور هستند که محصولات پروتئینی آنها مسئول جلوگیری از تقسیم سلولهای کنترلنشده میباشند. بهطور خاص، پروتئینهای BRCA1 و BRCA2 در بهبود آسیب به DNA و کنترل ژنهای دیگر دخیل هستند. علت اینکه جهش در این ژنها بهطور خاص به سرطان پستان و تخمدان مرتبط است، کاملاً مشخص نیست، اما ممکن است به هورمون استروژن مربوط باشد. سلولهای پستان و تخمدان برای تکثیر به هورمون استروژن وابستهاند و به تغییر مقادیر استروژن واکنش نشان میدهند (برای مثال، مقادیر استروژن تحت تاثیر چرخه قاعدگی و بلوغ است). تقسیم سریع ناشی از استروژن ممکن است منجر به افزایش جهش در این ژنها و رشد بعدی سرطان گردد (6) (7) (8).
سندرم لی– فرامینی
ژن: TP53
انواع سرطانهای مرتبط: سرطان پستان، سارکوم بافت نرم، استئوسارکوم (سرطان استخوان)، لوسمی، تومورهای مغزی، کارسینوم آدرنوکورتیکال (سرطان غدد فوق کلیه) و سرطانهای دیگر
ژن TP53 (یا ژن P53) یک پروتئین بسیار مهم سرکوبگر تومور را کپی میکند. این ژن در بسیاری از فعالیتهای جلوگیریکننده از تقسیم غیرقابل کنترل سلولی دخالت دارد. فعالیتهای مهم تنظیمشده توسط P53 عبارتند از: ترمیم DNA، مرگ سلولی (آپوپتوز) و کنترل چرخه سلولی. به این ژن “دروازهبان” میگویند بدین لحاظ که کار آن در نهایت محافظت بدن از تشکیل تومور است. جهشهای ارثی در TP53 باعث مستعد شدن فرد به انواع سرطان میشود، زیرا این ژن یکی از اصلیترین عوامل دفاعی بدن در برابر فعالیتهای سرطانزا است (9) (10) (11) (12).
سندرم کاودن (سندرم تومور هامارتوم P TEN)
ژن:TENP (همولوگ فسفاتاز و تنسین)
انواع سرطانهای مرتبط: پستان، تیروئید، آندومتر (پوشش رحم) و سرطانهای دیگر
PTEN نیز یک ژن سرکوبگر توموراست. درست مانند TP53 اگر PTEN هم معیوب باشد، سلولها ممکن است به تقسیم شدن ادامه دهند، حتی ممکن است تغییرات ایجادکننده سرطان رخ بدهند. بهطور خاص، محصول PTEN، پاسخ مثبت یا منفی سلولها به پیامهای تقسیم شدن و یا تحت آپوپتوز (نسخه خودکشی سلولی) قرار گرفتن را کنترل میکند. اگر تنظیم این پیامها غیرفعال شود، سلولها ممکن است بیوقفه و خارج از کنترل تقسیم شوند و تومور تشکیل شود. جهشهای PTEN مسئول سندرم کاودن که شامل شکل گرفتن بسیاری از هامارتومها که رشد غیربدخیمی هستند، میباشند. جهشها همچنین منجر به افزایش ریسک سرطان پستان، سرطان تیروئید و سرطان آندومتر (سرطان پوشش رحم) میشوند (13) (14) (15).
سندرم لینچ (سرطان کولورکتال غیرپولیپوز ارثی)
ژنها:MSH2 ،MLH1 ، MSH6،PMS2 ،EPCAM
انواع سرطانهای مرتبط: سرطانهای کولورکتال، آندومتر، تخمدان، لگن کلیوی، پانکراس، روده کوچک، کبد و مجرای صفراوی، معده، مغز و پستان
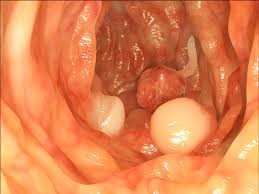
ژنهای درگیر در سندرم لینچ، ژنهای بازسازی نامتقارن DNA هستند. پروتئینهایی که توسط این ژنها کدگذاری شدهاند مسئول اصلاح اشتباهاتی هستند که هنگام کپی شدن (تکثیر) DNA بوجود آمدهاند. هنگامی که این ژنها معیوب باشند، پروتئینها بهدرستی قادر به بازسازی DNA نخواهند بود. اغلب سرطانهای مرتبط با سندرم لینچ رامیتوان با “بیثباتی ریزماهوارهها” شناسایی کرد. ریزماهواره اصطلاح توالی تکرارشونده DNA مانندCGCGCGCGG یا TATATATAT است. ژنوم انسانی از این نوع توالی تکرارشونده زیاد دارد. بیثباتی ریزماهوارهها یعنی جهشهایی که بهطور خاص در این توالیهای تکرارشوندهی DNA رخ میدهند. معمولا ًبرخی از تکرارها از بین رفته و یا اضافه میشوند (مثلاً CAGCAGCAG به CAGCAG تبدیل میشود).
تغییرات در توالیهای تکرارشونده میتوانند بر روی پایداری DNA تأثیر گذاشته و منجر به ایجاد انواع سرطانها شوند (6) (7) (8).
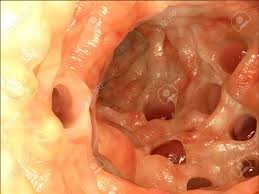
پولیپوز آدنوماتوز فامیلی
ژن: APC (کولیت پولیپوز آدنوماتوز)
انواع سرطانهای مرتبط: سرطان کولورکتال، تومورهای روده کوچک، مغز، معده، استخوان، پوست و بافتهای دیگر. همچنین با رشدهای (پولپهای) غیرسرطانی (خوشخیم) روده بزرگ و روده کوچک همراه است.
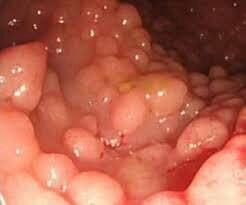
APC یک ژن سرکوبگر توموراست که چگونگی تقسیم سلولی، چگونگی چسبیدن سلولها به یکدیگر و چگونگی حرکت سلولی را کنترل میکند. این ژن همچنین در تشخیص آسیب DNA سهیم است و با سایر پروتئینهای دخیل در ارتباط بین سلولها همراهی میکند. بسیاری از جهشهای مختلف در APC بهعنوان علت ایجاد پولیپوز آدنوماتوز فامیلی شناخته شدهاند، یک بیماری که با پیدایش صدها پولیپ توأم است. بسیار محتمل است که حداقل یکی از پولیپها در برخی مواقع در طول زندگی بیمار بتواند سرطانی شود. پروتئین معیوب APC نیز میتواند منجر به ایجاد تومورهای دسموئید که تومورهای خوشخیم ضخیم بافت همبند هستند، بشود (16) (17) (18) (19).
رتینوبلاستوم
ژن: RB1 (رتینوبلاستوم)
انواع سرطانهای مرتبط: سرطان چشم (سرطان شبکیه)، پینهآلوم (سرطان غده صنوبری)، استئوسارکوم، ملانوم و سارکوم بافت نرم
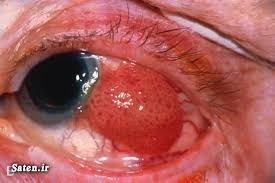
ژن RB1 که یک ژن سرکوبگر است، پروتئین Rb را کد میکند. پروتئین Rb مسئول متوقف کردن تقسیم سلولی در شرایط نامطلوب است (بهطور مثال در هنگام آسیب DNA که بایستی بازسازی شود، یا زمانی که سلول بهنوعی تحت فشار است). این ژن در کنترل سایر پروتئینهایی که در تکثیر DNA، آپوپتوز و بلوغ سلولی (تمایز) دخیل هستند، نقش دارد. هنگامی که جهشی در ژن RB1 رخ دهد، پروتئین Rb ممکن است فعالیت نکند، در این صورت رشد سلولی بیقاعده خواهد شد.
به دلایلی که بهطور کامل مشخص نیست، تغییرات در RB1 باعث ایجاد سرطان در چشم بهویژه در شبکیه میشود. زمانی که یک کپی جهشیافته از ژن رتینوبلاستوم به ارث برده شود (باعث ایجاد سرطانهایی به نام رتینوبلاستوم ژرمینال یا فامیلیال شود)، ژن جهشیافته در هر سلول بدن یافت میشود و فرد را بهشدت به انواع دیگر سرطان، در درجه اول سرطان غده صنوبری، استخوانها، بافت نرم و پوست، حساس میکند (20) (21) (22) (23) (24).
مولتیپل اندوکرین نئوپلازی تیپ 1 (سندرم ورمر)
ژن: MEN1
انواع سرطانهای مرتبط: تومورهای درونریز پانکراس و (معمولاً خوشخیم) تومورهای پاراتیروئید و غده هیپوفیز
MEN1 یک پروتئین سرکوبگر تومور به نام منین را کپی میکند. عملکرد دقیق منین ناشناخته است، اما بهنظر میرسد که در تنظیم تقسیم سلولی، بازسازی DNA و آپوپتوز دخیل باشد. بیش از هزار جهش مختلف درژن MEN1 موجب ایجاد مولتیپل اندوکرین نئوپلازی تیپ 1 میشوند. تیپ 1 ژن MEN در رشد تومور داخل غدد درونریز (غدد تولیدکننده هورمون) مداخله میکند. غدد اندوکرینی که غالباً تحت تأثیر مولتیپل نئوپلازی تیپ 1 هستند عبارتند از غده پاراتیروئید، غده هیپوفیز و پانکراس.
معمولاً جهش در MEN1 منجر به تولید نسخه کوتاهشده پروتئین منین میشود که ناپایداراست و بهراحتی تجزیه میشود. وقتی چنین چیزی اتفاق میافتد، یک کپی از ژن MEN1، پروتئین عملکردی منین را تولید نمیکند. اگر جهش در نسخه دوم (که در غدد درونریز عادی است، اگرچه دلیل آن ناشناخته است)، رخ دهد، سلول نمیتواند بههیچوجه منین مناسبی را تولید کند و منجر به تقسیم غیرقابل کنترل سلول و سرطان میگردد (25) (26) (27) (28) (29) (30).
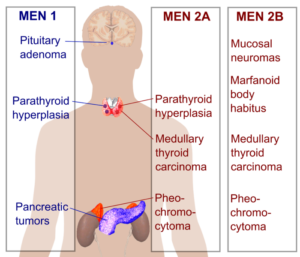
مولتیپل اندوکرین نئوپلازی تیپ 2
ژن:RET
انواع سرطانهای مرتبط: سرطان تیروئید مدولار و فئوکروموسیتوم (تومور خوشخیم غده فوق کلیه)
ژن RET یک پروتوانکوژنی است که در پیامرسانی سلولی دخالت دارد و غشای سلولی را دربر میگیرد و بهعنوان گیرندهای برای پیامهایی که به پاسخ سلولها به تغییرات محیطی اطرافشان کمک میکنند، عمل مینماید. MEN2 به سه زیرگونه تقسیم میشود: MEN2A ,MEN2B و کارسینوم تیروئید مدولاری فامیلی (FMTC). بیشتر جهشها در ژن RET که باعث ایجاد MEN2 میشوند جهشهای بسیار کوچک (نقطهای) هستند که باعث تغییر در فقط یک اسیدآمینه پروتئین میشوند. بسیاری از این جهشها با سرطان تیروئید مدولاری ارثی (فامیلی) مرتبط هستند (27) (28) (30) (31) (32).
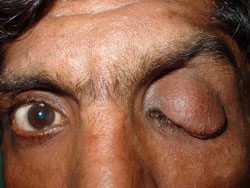
سندرم فون هیپل– لیندو
ژن: VHL
انواع سرطانهای مرتبط: سرطان کلیه و تومورهای متعدد غیر سرطانی از جمله فئوکروموسایتوم
ژن VHL به همراه سایر پروتئینها کمپلکس VCB- CUL2 را تشکیل میدهند. این کمپلکس باعث میشود که سایر پروتئینهای درون سلول، زمانی که آسیب دیدهاند و یا دیگر نیازی به آنها نباشد، شکسته شوند. یکی ازتارگتهای این کمپلکس، فاکتور القاءشده با هیپوکسی 2-alpha(HIF2a) است. HIF2a پاسخ بدن به تغییرات سطح اکسیژن را با کنترل تقسیم سلولی و تشکیل رگهای خونی جدید و گلبولهای قرمز انطباق میدهد.
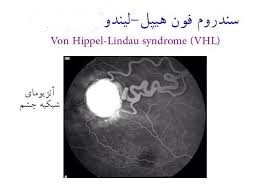
وقتی که سطح اکسیژن نرمال باشد، کمپلکس VCB- CUL2، فاکتور HIF2a را متوقف میکند. زمانی که VHL جهش پیدا کند، کمپلکس VCB- CUL2 بهدرستی عمل نخواهد کرد و نمیتواند HIF2a یا پروتئینهای دیگری که آسیب دیدهاند و یا موردنیاز نیستند را تخریب کند، در این صورت ممکن است HIF2a باعث تحریک تقسیم بیش ازحد سلولی و تشکیل عروق خونی شود که میتواند منجر به ایجاد تومور و کیست که هر دو مشخصه سندرم فون هیپل- لیندو هستند، گردد (33) (34) (35) (36) (37).
منابع:
- gov website: Genetic testing fact sheet. Reviewed April 2013. [http://www.cancer.gov/about-cancer/causesprevention/genetics/genetic-testing-fact-sheet]
- Larki P, Khoshbakht S, Rezaei N. Genetics of neoplasia: inherited monogenic defects associated with cancers. Acta Med Iran. 2014;52(1):91-2. [PUBMED]
- Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008 Nov 13;359(20):2143-53. [PUBMED]
- NIH Genetics Home Resource: Genetic Testing. Published August 3, 2015 [ http://ghr.nlm.nih.gov/handbook/testing/procedure]
- NCI – Genetic Testing for Hereditary Cancer Syndromes Reviewed April 2013 [http://www.cancer.gov/about-cancer/causesprevention/genetics/genetic-testing-fact-sheet]
- b.Tiwari AK, Roy HK, Lynch HT. Lynch Syndrome in the 21st Century: Clinical Perspectives. QJM. 2015 Jul 29. pii: hcv137. [Epub ahead of print] [PUBMED]
- b.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010 Jun;138(6):2073-2087 [
- b.NIH Genetics Home Reference: Lynch Syndrome Published 08-03-1025 [http://ghr.nlm.nih.gov/condition/lynchsyndrome]
- Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, Gauthier-Villars M, Stoppa-Lyonnet D, Consolino E, Brugières L, Caron O, Benusiglio PR, Bressac-de Paillerets B, Bonadona V, Bonaïti-Pellié C, Tinat J, BaertDesurmont S, Frebourg T. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol. 2015 Jul 20;33(21):2345-52. Epub 2015 May 26. [PUBMED]
- Mork ME, You YN, Ying J, Bannon SA, Lynch PM, Rodriguez-Bigas MA, Vilar E. High Prevalence of Hereditary Cancer Syndromes in Adolescents and Young Adults With Colorectal Cancer. J Clin Oncol. 2015 Jul 20. pii: JCO.2015.61.4503. [Epub ahead of print] [PUBMED]
- Merino D, Malkin D. p53 and hereditary cancer. Subcell Biochem. 2014;85:1-16. [PUBMED]
- Toss A, Tomasello C, Razzaboni E, Contu G, Grandi G, Cagnacci A, Schilder RJ, Cortesi L. Hereditary Ovarian Cancer: Not Only BRCA 1 and 2 Genes. Biomed Res Int. 2015;2015:341723. Epub 2015 May 17. [PUBMED]
- From the NIH Genetics Home Reference. Published August 3, 2015 [http://ghr.nlm.nih.gov/gene/PTEN]
- Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: Multiple Functions in Human Malignant Tumors. Front Oncol. 2015 Feb 16;5:24. [PUBMED]
- Haas NB, Nathanson KL. Hereditary kidney cancer syndromes. Adv Chronic Kidney Dis. 2014 Jan;21(1):81-90 [PUBMED]
- NIH Genetics Home Reference: APC Reviewed March 2013, Published 8-3-2915 [http://ghr.nlm.nih.gov/gene/APC]
- NIH Genetics Home Reference. Familial Adenomatous Polyposis Reviewed October 2013, Published August 3, 2015 [ http://ghr.nlm.nih.gov/condition/familial-adenomatous-polyposis]
- Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009 Oct 12;4:22. [PUBMED] Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer. 2003 Dec 12;2:41. [PUBMED]
- NIH Genetics Home Reference: Retinoblastoma. Reviewed August 2009, Published August 3, 2015 [ http://ghr.nlm.nih.gov/condition/retinoblastoma]
- Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006 Aug 28;25(38):5220-7. [PUBMED]
- Lohmann DR, Gallie BL. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2000 Jul 18 [updated 2013 Mar 28] [PUBMED]
- Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011 Aug 10;476(7359):163-9. [
- Field M, Shanley S, Kirk J. Inherited cancer susceptibility syndromes in paediatric practice. J Paediatr Child Health. 2007 Apr;43(4):219-29. [PUBMED]
- NIH Genetics Home Resource: MEN1 Reviewed August 2013, Published August 3, 2015 [http://ghr.nlm.nih.gov/gene/MEN1]
- Gaztambide S, Vazquez F, Castaño L. Diagnosis and treatment of multiple endocrine neoplasia type 1 (MEN1). Minerva Endocrinol. 2013 Mar;38(1):17-28. [PUBMED]
- b.Carney JA. Familial multiple endocrine neoplasia: the first 100 years. Am J Surg Pathol. 2005 Feb;29(2):254-74. [
- b.Anik A, Abaci A. Endocrine cancer syndromes: an update. Minerva Pediatr. 2014 Dec;66(6):533-47. Epub 2014 Sep 22. [PUBMED]
- Sovrea AS, Dronca E, Galatâr M, Radian S, Vornicescu C, Georgescu C. Diagnostic correlation between RET protooncogene mutation, imaging techniques, biochemical markers and morphological examination in MEN2A syndrome: case report and literature review. Rom J Morphol Embryol. 2014;55(2):389-400. [PUBMED]
- b.NIH Genetics Home Reference – Multiple Endocrine Neoplasia. Reviewed August 2013, Published August 3, 2015 [ http://ghr.nlm.nih.gov/condition/multiple-endocrine-neoplasia]
- Sovrea AS, Dronca E, Galatâr M, Radian S, Vornicescu C, Georgescu C. Diagnostic correlation between RET protooncogene mutation, imaging techniques, biochemical markers and morphological examination in MEN2A syndrome: case report and literature review. Rom J Morphol Embryol. 2014;55(2):389-400. [PUBMED]
- Jessica Marquard, MS, LGC and Charis Eng, MD, PhD, FACP. Multiple Endocrine Neoplasia Type 2, Synonyms: MEN 2, MEN2 Syndrome NCBI Gene Reviews – Initial Posting: September 27, 1999; Last Update: June 25, 2015. [ http://www.ncbi.nlm.nih.gov/books/NBK1257/]
- Kaelin WG Jr. “Molecular basis of the VHL hereditary cancer syndrome.” Nat Rev Cancer (2002). 2(9):673-82. [PUBMED]
- Cowey CL, Rathmell WK. VHL gene mutations in renal cell carcinoma: role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep. 2009 Mar;11(2):94-101. [PUBMED]
- Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de Bruine AP. VHL and HIF signalling in renal cell carcinogenesis. J Pathol. 2010 Jun;221(2):125-38. doi: 10.1002/path.2689. [PUBMED]
- Genetics Home Reference. National Institutes of Health. U.S. Department of Health and Human Services. Reviewed August 2012, Published August 2015 [http://ghr.nlm.nih.gov/gene/VHL]
- Shahzad H, Kehar SI, Ali S, Tariq N. Expression of Von Hippel – Lindau (VHL) gene mutation in diagnosed cases of renal cell carcinoma. Pak J Med Sci. 2014 Jul;30(4):880-5. [PUBMED]
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام