جداسازی و تخلیص اسیدهای نوکلئیک
دکتر مهدی فصیحی رامندی (عضو هیئت علمی دانشگاه علوم پزشکی بقیه ا…(عج))
زهرا کریمی (مرکز تحقیقاتی زیست سلول پژوهان تدبیر)
دکتر رضا میرنژاد (استاد دانشگاه)
اولین قدم در ژنتیک، جداسازی و تخلیص DNA و RNA است. این کار دارای مراحلی است که در ادامه به آن اشاره میشود.
DNA را میتوان از منابع مختلفی از جمله خون کامل حاوی ضد انعقاد، لخته خون، گلبول سفید، رده سلولی، میکروارگانیسمهای مختلف و … جدا نمود. بهمنظور استخراج DNA از خون کامل بهتر است از خون تازه استفاده شود. اگر امکان استخراج سریع وجود نداشته باشد، نمونه بهصورت کامل و استریل در دمای اتاق نگهداری میشود. اگر این تأخیر بیش از یک هفته باشد نمونه را در میکروتیوبها توزیع کرده و در دمای70-/20- حداکثر به مدت یک سال نگهداری میکنند. برای نگهداری طولانی مدت نمونهها میتوان لنفوسیتهای خون را جدا کرده و در محیط حاوی DMSO[1] درون نیتروژن مایع قرار داد.
نکات قابل توجه
- احتمال آلودگی و عفونت در نمونههای خون مورد استفاده وجود دارد؛ بنابراین هنگام کار با این نمونهها نکات ایمنی را رعایت کنید.
- ضدانعقاد EDTA یا سیترات برای کار با DNA مناسب است، اما هپارین مهارکننده PCR است. نمونههایی که بیش از یک هفته از تهیه آنها گذشته است، از لحاظ کمّی و کیفی DNA خوبی نمیدهند؛ مگر اینکه فریز شده باشند. میزان DNA حاصل، بستگی به تعداد گلبولهای سفید نمونه دارد. هر میلیلیتر از خون کامل حاوی حدود 10000 گلبول سفید بوده و از آن gµ100 نمونه DNA بدست میآید.
- هنگام تخلیص باید دقت شود تا آلودگی با دیگر نمونهها، DNA و محصولات PCR بهوجود نیاید، بدین منظور باید دو اتاق جدا تحت عنوان Pre-PCR و Post-PCR تعبیه شود. در اتاق Pre-PCR کارهایی از قبیل جداسازی DNA و PCR انجام شده و در اتاق دیگر بر روی محصول PCR کار میشود. وسایل و مواد از قبیل روپوش، سمپلر و بافرهای هر اتاق باید مجزا و مشخص باشد. کارهای اتاق Pre-PCR زیر هود انجام میشود.
- هنگام کار حتماً از دستکش استفاده شود. از لولهها و میکروتیوبهای یک بار مصرف و وسایل شیشهای قابل اتوکلاو استفاده گردد. از مواد پلاستیکی (سرسمپلر، لوله، میکروتیوب) با جنس پلیپروپیلن استفاده شود. مواد با جنس نامرغوب میتواند DNA را به خود جذب کند.
مراحل جداسازی اسید نوکلئیک
شکستن سلول: از آنجا که DNA و RNA در داخل سلول قرار دارند، جهت جداسازی آنها ابتدا باید سلول را شکست. برای این کار در مورد هر سلول باید روش خاص آن را بهکار برد؛ برای مثال سلولهای جانوری را معمولاً با استفاده از شوک اسمزی و یا دترجنتهای یونی مثل SDS (سدیم دودسیل سولفات) و سلولهای گیاهی را به دلیل داشتن دیواره سخت پکتینی و سلولزی، با استفاده از آنزیمهای پکتیناز و سلولاز میشکنند. شکستن دیواره باکتریها نیز با توجه به وابستگی آنها به دو گروه عمده گرم مثبت و گرم منفی، با تکنیکهای خاصی صورت میگیرد.
باکتریهای گرم مثبت لایه پپتیدوگلیکان ضخیمی در دیواره سلولی خود دارند، بنابراین جهت شکستن دیواره آنها از آنزیم لیزوزیم و مدت زمان نسبتاً زیاد استفاده میکنیم.
باکتریهای گرم منفی در لایه بیرونی خود یک غشای خارجی نیز دارند که این غشاء مانع از عمل لیزوزیم بر روی دیواره آنها میشود، بنابراین در این گروه از باکتریها علاوه بر لیزوزیم از EDTA (اتیلن دیآمین تترا استیک اسید) استفاده میشود. EDTA باعث حل شدن غشای خارجی شده و در نتیجه دیواره سلولی در معرض لیزوزیم قرار میگیرد.
لازم به ذکر است که به دلیل شکنندگی رشتههای RNA و DNA، شکستن سلولها باید در داخل بافر انجام گیرد. این بافر شامل HCl Tris[2]– و SDS است. SDS دو خاصیت دارد: یکی باعث حل شدن چربیها میشود و دیگر اینکه با اتصال به DNA باعث پایداری آن میگردد.
رسوب دادن DNA و RNA: با شکسته شدن سلول، محتویات آن شامل DNA و RNA پروتئین، چربی، قند و … آزاد میشود. در مرحله بعد باید DNA و RNA را خالص کرد. برای این کار از الکل استفاده میشود. الکل سرد را به سوسپانسیون حاصل از شکستن سلول اضافه کرده و در فریزر قرار میدهند. در این مرحله DNA و RNA رسوب میکنند.
جداسازی DNA و RNA: برای این کار بهطور معمول از میله شیشهای استفاده میشود؛ به این صورت که میله را بهآرامی داخل لوله فرو برده و میچرخانند. رشتههای DNA و RNA به دور میله شیشهای میچسبند، سپس میله را بهآرامی خارج کرده و در یک لوله دیگر محتوی بافر فرو برده و تکان میدهند تا DNA و RNA دوباره در بافر حل شوند. این مرحله باید با ملایمت صورت گیرد تا DNA و RNA نشکنند.
رسوب پروتئین: DNA و RNA حاصل از مرحله قبل هرچقدر هم که خالص باشند، ولی باز هم حاوی کمی پروتئین خواهند بود. در این مرحله برای خالص کردن DNA و RNA باید پروتئینها را رسوب دهیم. برای این کار از فنل و یا محلول غلیظ نمکهای مختلف استفاده میکنند. این مواد باعث تغییر شکل پروتئینها (دناچوره شدن) و در نتیجه رسوب آنها میشود.
جدا کردن DNA و RNA از یکدیگر: در این مرحله با توجه به کاری که مدنظر است DNA و RNA جدا میشوند. اگر هدف بررسی DNA باشد با استفاده از آنزیم RNase، RNAها را تجزیه میکنند و اگر بخواهند RNA را بررسی کنند، از آنزیم DNase استفاده میکنند.
استخراج DNA به روش Salting out
- ايزوپروپانول (موجب رسوب DNA میشود و بايد در 4 درجه سلسیوس نگهداري شود)
- اتانول 70% (موجب رسوب DNA میشود و بايد در 4 درجه سلسیوس نگهداري شود)
- SDS 10% (اين محلول پروتئينها را حذف كرده و آنها را رسوب ميدهد)
- نگهداري در دماي اتاق
- آب مقطر سرد
- TE براي حل كردن DNA (ميتوان DNA را در آب مقطر استريل نيز حل كرد، اما بافر TE براي نگهداري طولاني مدت مناسبتر است، زيرا pH اسيدي موجب دپورينه شدن DNA و pH قليايي سبب تخريب DNA میشود. همچنين EDTA با حذف كاتيونهاي دو ظرفیتی، آنزيم DNAase را غیرفعال ميكند).
– 10 mM Tris-HCL
– 0.2mM Na2EDTA
pH اين بافر بايد به 7/4 برسد و بعد از اتوكلاو در 4 درجه سلسیوس نگهداري شود.
- بافر ليز هسته
- -10 mM Tris-HCL
– 400mM NaCl
– 0.2mM Na2EDTA
pH اين بافر بايد به 8/4 برسد و بعد از اتوكلاو در 4 درجه سلسیوس نگهداري شود.
- محلول پروتئيناز K (آنزيم DNAase را غیرفعال ميكند)
- 1mg/ml محلول پروتئينازk (اين آنزيم در 20- درجه سلسیوس نگهداري میشود).
ابتدا 5 میلیلیتر خون حاوي EDTA را به مدت 10 دقيقه در g1000 در دماي صفر درجه سلسیوس سانتريفيوژ كرده و محلول رويي را بهآرامی جدا كنيد.
بر روي رسوب باقيمانده 10 میلیلیتر آب مقطر استريل با دماي 4 درجه سلسیوس ريخته و خوب مخلوط كنيد، سپس 10 دقيقه در دماي صفر درجه سلسیوس در g1000 سانتريفيوژ كرده و پس از آن مايع رويي آن را دور بريزيد. اين عمل را چندين بار تكرار كنيد تا مايع رويي کاملاً شفاف و رسوب بدون رنگ باشد.
بر روي رسوب باقيمانده در انتهاي لوله، 4/5 میلیلیتر بافر ليزكننده حاوي 10 ميليمــــول Tris-HCL و 150 ميليمول EDTA، 10 ميليمول NaCL با pH=7.5 ريخته و به آن 20 ميكروليتر پروتئيناز K (mg/ml10) و 250 ميكروليتر SDS 10% اضافه كنيد، سپس محلول حاصل را به مدت 24 ساعت در دماي 37 درجه سلسیوس قرار دهيد تا عمل هضم انجام پذيرد. پس از هضم سلولها، جهت رسوب كردن پروتئين 1/8 میلیلیتر محلول اشباع كلريد سديم 5M بر روي آن اضافه كرده و بهآرامی تكان دهيد. اين محلول را به مدت 25 دقيقه در g1000 در دماي 21 درجه سلسیوس سانتريفيوژ نماييد.
در صورتيكه محلول رويي شفاف نبود، محلول رويي را جدا كرده و به آن مجدداً 500 ميكروليتر كلريد سديم اشباع افزوده و سانتريفيوژ كنيد تا کاملاً شفاف گردد. محلول شفاف رويي را جدا كرده و به يك لوله تميز انتقال دهيد، سپس بهآرامی بر روي آن 5 میلیلیتر ايزوپروپانول خنك (4 درجه) اضافه كنيد تا كلاف DNA مشاهده شود. كلاف را بهآرامی با سمپلر برداشته و به يك ميكروتيوب منتقل كنيد، سپس محلول اضافي روي آن را پس از ميكروفيوژ كردن برداشته و 400 میلیلیتر اتانول 70% به آن اضافه كنيد.
الكل اضافه را مجدداً پس از ميكروفيوژ كردن با كمك سمپلر برداشته و ميكروتيوب را در 45 درجه سلسیوس قرار دهيد تا الكل آن تبخير گردد. سپس به ميكروتيوب حاوي DNA، 200 ميكروليتر بافر TE حاوي 10 ميليمولار Tris-HCL و يك ميليمولار EDTA با pH=7.4 اضافه كنيد تا DNA در آن حل شود. ميكروتيوب را به مدت يك شب در دماي آزمايشگاه قرار داده و سپس در دماي 4 درجه سلسیوس در يخچال نگهداري كنيد. اگر فعلاً كاري بر روي DNA انجام نميدهيد، بهتر است آن را در 20- درجه سلسیوس نگهداري نماييد.
lµ10 از محلول را به حجم lµ1000 رسانده و OD آن را در طولموج nm260 و nm280 بررسي كنيد. نسبت را محاسبه كنيد. (خوانش OD توسط دستگاه نانودراپ)
نكات قابل توجه:
ضريب رقت در هنگام محاسبه غلظت فراموش نشود.
OD=1 در طولموج nm260 معادل µg50 از DNA دو رشتهاي (ds DNA) میباشد.
OD=1 در طولموج nm260 معادل µg50 از DNA تك رشتهاي (ss DNA) میباشد.
بهترين حالت براي نسبت OD260 به OD280، عددي در حدود 1/8 میباشد. (همانطور که در شکل زیر مشاهده میشود)
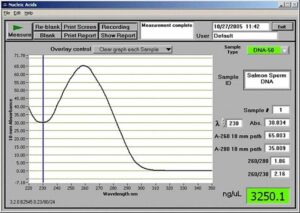
اگر اين نسبت بيشتر از 1/8 باشد، آلودگي با RNA وجود دارد و اگر از 1/8 كمتر باشد، DNA با پروتئين آلوده است.
براي اندازهگيري پروتئين موجود در نمونه برحسب mg/ml ميتوان از فرمول زير استفاده كرد:
استخراج DNA از خون به روش فنل- كلروفرم
محلولهاي موردنیاز:
1-Reticolocyte salin
0.13M NaCl
5mM KCl
7.4mM MgCl2
2- Lysis solution (10X)
0.77M NH4 Cl
0.046M KHCO3
3- TE Buffer
10mM Tris-HCl (pH=8.0)
1mM EDTA (pH=8.0)
4- Lysis solution
100mM NaCl
25mM EDTA (pH=8.0)
5- SDS (10%)
6- NaCl (5M)
7- Ammonium Acetate (10M)
روش كار:
- 10 ميليليتر خون حاوي EDTA را داخل لوله فالكون 50 سيسي ريخته و 20-10 سيسي از محلول Reticolocyte salin را به آن اضافه كنيد.
- به مدت 1 دقيقه با دور g1000(تقریباً 3000 دور در دقيقه) سانتريفيوژ نماييد.
- بهآرامی پلاسما را جدا كنيد. دقت شود به لايه بافيكوت دست نزنيد.
- لوله فالكون را تا حجم ml50 با محلول ليز (x2) پر كرده و به مدت ده دقيقه مخلوط كنيد تا گلبولهاي قرمز ليز شوند.
- محلول فوق را 10 دقيقه با دور g1000 سانتريفيوژ نماييد.
- در اين هنگام كه رسوب گلبولهاي سفيد در ته لوله تشكيل ميگردد، بهآرامی و با سمپلر مايع رويي را برداريد.
- به رسوب سلولي مواد زير را اضافه كنيد:
- – محلول ليز ml10
SDS (10%) ml0/5 –
– آنزيم K پروتئيناز (mg/ml20) lµ20
- سپس محلول حاصل را خوب مخلوط كرده، حداقل 4 ساعت (يا over night) در 37 درجه سلسیوس انكوبه كنيد.
- سپس به اندازه نصف حجم محلول حاصل، از فنل متعادلشده با محلول تريس يك مولار (pH=8) و نصف ديگر از كلروفرم بريزيد.
- محلول را به مدت 10 دقيقه با دور g3000 سانتريفيوژ كرده، لايۀ رويي كه لايه آبي و حاوي اسيد نوكلئيك است را بهآرامی جدا كرده و در ظرف ديگري بريزيد.
- مرحله افزودن فنل کلروفرم را بار ديگر تكرار نماييد.
- دوباره محلول رويي را خارج نموده و در لوله تميز بريزيد. سپس به اندازه نصف حجم محلول، به آن كلروفرم اضافه كرده و مطابق مرحله قبل سانتریفیوژ نماييد.
- به محلول رويي جداشده به اندازه نصف حجم آن استات آمونيم 10 مولار و دو برابر حجم آن اتانول 96 درجه اضافه كنيد. با سر و ته كردن لوله ميتوان كلاف DNA را مشاهده كرد.
- لوله را 5 دقيقه با دور g1000 سانتريفيوژ نماييد.
- بهآرامی محلول رويي را خارج كرده و رسوب را با اتانول 70 درجه بشوييد.
- لوله را به مدت حداقل 10 دقيقه در حرارت اتاق قرار داده تا کاملاً اتانول تبخير شود. سپس رسوب را در 1-0/5 ميليليتر آب مقطر يا بافر TE حل كرده و در 20- درجه سلسیوس نگهداري نماييد.
تخليص DNA از خون به روش جوشاندن
محلولهاي موردنیاز:
- بافر R (بافر ليز)
- ساكاروز 0/32 مول
- تريس-HCl 10 ميليمولار
- كلريد منيزيم 5 ميليمولار
- تريتون (X100) 1%
محلول 1:
NaOH mM 50
محلول 2:
تريس-HCl 1 مولار (7/5pH=)
روش كار:
- مقدار ml0/5 خون را در يك ميكروتيوب ml1/5 ريخته و به آن يك ميليليتر از بافر R اضافه كرده، چند بار سروته كنيد. به مدت يك تا دو دقيقه با دور rpm10000 سانتريفيوژ كنيد. محلول رويي را دور بريزيد
- مقدار lµ250 از بافر R را به رسوب اضافه كرده، ورتكس كنيد و سپس lµ750 ديگر از بافر R اضافه كرده و بعد از مخلوط كردن كامل، مشابه مرحلۀ فوق سانتريفيوژ نماييد. اين كار را آنقدر ادامه دهيد تا رسوب کاملاً سفيد شود.
- lµ100 از محلول سود (NaOH) را به رسوب اضافه نموده و مخلوط كنيد. سپس به مدت 20 دقيقه بجوشانيد (بعد از اين مرحله هيچ رسوبي نبايد در تيوب باقي بماند).
- lµ20 از محلول تريس يك مولار را به محلول فوق را اضافه كرده، مخلوط كنيد.
- دقیقاً 30 ثانيه سانتريفيوژ كنيد.
- براي حذف پروتئين ميتوانید lµ5 پروتئيناز K را اضافه كرده و بعد از مخلوط كردن به مدت يك ساعت در دماي 55 درجه سلسیوس قرار دهيد. در اين مدت هر 5 دقيقه يك بار ميكروتيوب را سروته نماييد.
- مايع رويي حاوي DNA میباشد كه برداشته شده و در 20- درجه سلسیوس نگهداري شود.
نكته: از معايب اين روش خرد و قطعهقطعه شدن DNA میباشد، بنابراين DNA حاصل براي PCR قطعات بزرگ DNA مناسب نیست.
تخليص DNA از خون به روش Salazar
محلولهاي موردنیاز:
- بافر 1 (بافر ليز گلبول قرمز)
- تريس-HCl mM10 (pH=8)
- كلريد پتاسيم (KCl) mM10
- كلريد منيزيم (MgCl2) mM10
- EDTA mM 2 (pH=8)
- تريتون X 100 (2/5%)
- بافر 2 (لیز گلبول سفيد)
- تريس-HCl mM10 (pH=8)
- كلريد پتاسيم (KCl) mM10
- كلريد منيزيم (MgCl2) mM10
- EDTA mM 2 (pH=8)
- كلريد سديم (NaCl) 0/4 مولار
- SDS g/L10
- كلريد سديم 5 مولار
- بافر TE(محلول نگهدارنده TE)
- تريس-HCl mM10(pH=7.5)
- EDTA mM1
روش كار:
- يك سيسي از خون EDTAدار را داخل ميكروتيوب ريخته و 5 دقيقه با g1200 سانتريفيوژ كنيد.
- مايع رويي را دور بريزيد.
- گلبول قرمز را با ml1 از بافر 1 ليز كنيد.
- بعد از سانتريفيوژ، رسوب را دوباره با بافر 1 شستشو دهيد. اين كار را آنقدر ادامه دهيد تا رسوب کاملاً سفيد شود.
- رسوب حاصل را كه کاملاً سفيد رنگ است با lµ220 از بافر 2 ليز كنيد.
- محلول حاصل را 15 دقيقه در 56 درجه سلسیوس انكوبه كنيد.
- در اين مرحله lµ100 از محلول كلريد سديم 5 مولار را اضافه كرده تا پروتئينها رسوب داده شوند.
- ميكروتيوب را به مدت 10 دقيقه سانتريفيوژ نموده و مايع رويي را كه حاوي DNA است، بهآرامی با سمپلر جدا كنيد.
- دو برابر حجم برداشته شده از مرحله قبل، اتانول 96 درجه اضافه كنيد و به مدت 15 دقيقه در كنار يخ يا در 20- درجه سلسیوس قرار دهيد تا كلاف DNA را مشاهده نماييد.
- ميكروتيوب را 10 دقيقه با 10000 دور در دقيقه سانتريفيوژ كرده و رسوب را دو بار با اتانول 70% شستشو دهيد.
- رسوب حاصل را پس از تبخير كامل اتانول در lµ100 از بافر TE حل كرده و در شرايط مناسب نگهداري نماييد.
استخراج به روش [3]P.C.I
هدف از اين كار حذف پروتئینهای موجود در محلول حاوي DNA میباشد. فنل (فنل متعادلشده با pH بيشتر از 7/5) موجب دناتوده شدن پروتئینها میشود و كلروفرم نيز بقاياي فنل را از محلول واكنش حذف ميكند.
- همحجم محلول حاوي DNA، فنل اضافه كنيد و مدت 5 دقيقه سانتريفيوژ كنيد (فنل متعادلشده بهعلت داشتن ماده آنتیاکسیدان 8-hydroxyquinoline، زرد رنگ است و روي آن با يك لايه بافر تريس پوشانيده شده است. هنگام استفاده از فنل زير بافر استفاده كنيد)
- مايع رويي (مايع شفاف) را به لوله تميز ديگري انتقال دهيد (فنل در زير قرار ميگيرد و پروتئینهای دناتوره شده بين دو لايه قرار میگیرند).
- همحجم آن از مخلوط كلرفرم- ايزوآميل الكل (1:24) اضافه كنيد (ايزو آميل الكل يك ماده Anti foamاست و مانع كف كردن محلول میشود) و مانند مرحله قبل ادامه دهيد.
- مايع رويي را به لوله تميز ديگر انتقال دهيد و يا مانند روش زير میتوان عمل نمود:
- مخلوط فنل- كلرفروم- ايزو آميل الكل(به نسبت 1:24:25) تهيه كنيد.
- حجم مساوي از مخلوط C.I به محلول حاوي DNA اضافه كنيد و 5 دقيقه سانتريفيوژ كنيد.
- محلول رويي را به لوله تميز ديگر انتقال دهيد و همحجم آن I اضافه كنيد و 5 دقيقه سانتريفيوژ كنيد.
- محلول رويي را به لوله تميز ديگر انتقال دهيد (محلول حاوي DNA است)
- اگر هنوز بوي فنل از آن متصاعد میشود مرحله 7 را تكرار كنيد.
- DNA را با الكل رسوب دهيد (تغليظ DNA).
رسوب DNA با الكل (تغليظ DNA)
هدف از اين كار احياي DNA بعد از استخراج و يا بعد از استخراج P.C.I. میباشد و در مواقعي نيز كه DNA خيلي رقيق باشد بدين وسيله آن را تغليظ میکنیم.
- حجم محلول حاوي DNA را تعيين كنيد (تخمين بزنيد).
- استات سديم با غلظت نهايي 3M را به آناضافه كنيد.
- مقدار دو برابر حجم آن الكل مطلق سرد (20- يا 70-) اضافه نمائید.
- مدت 10 دقيقه با 12000g سانتريفيوژ كنيد. رسوب سفيد رنگ را میتوان در ته لوله مشاهده نمود (لازم به ذكر است كه DNA خالص تقریباً بیرنگ است).
- با وارونه كردن لوله، مايع رويي را حذف كنيد و لوله را بهصورت وارونه روي كاغذ جاذبالرطوبه قرار دهيد تا آب آن حذف شود.
- مقدار 100 ميكرو ليتر الكل 70 درجه به آن اضافه كنيد و آن را ورتكس كنيد تا رسوب DNA از ته لوله كنده شده و شناور گردد (اگر DNAبزرگ باشد ورتكس موجب شكسته شدن آن میشود).
- مدت 5 دقيقه آن را سانتريفيوژ كنيد (شستن DNA).
الكل را خالي كرده و DNA را در 70 درجه خشککنيد (چنانچه در اين مرحله DNA خوب خشك نشود و الكل همراه آب داخل آن بماند، بعداً براي كار كردن با آنزیمها دچار اشكال میشوید). DNA را در مقدار دلخواه بافر (آب) حل كنيد.
احياي DNA از ژل آگارز
هدف از اين قسمت كسب مهارت براي احياي DNA از روي ژل آگارز میباشد. اين تكنيك در بيولوژي مولكولي و مهندسي ژنتيك اهميت زيادي دارد و در همسانهسازی ژن و انتقال ژن استفاده میشود. لازم به ذكر است كه از طرق مختلف اين كار انجام میشود؛ ماننــــــــــــــد
Freeze- Squeeze ,paper elution ,Electro elution و… ولي در اینجا به توضیح دو روش بسنده میشود:
الف) احياي DNA از روی ژل توسط كاغذ (Paper elution)
- DNAموردنظر را با ژل آگاروز 2-1% الكتروفورز كنيد و اجازه دهيد تا نوارهاي DNA بهخوبی از همديگر تفكيك شوند.
- با نور UV (طولموج 254 نانومتر موجب آسیب DNA میشود و بهتر است از طولموج 312 نانومتر استفاده گردد) نوارهايموردنظر را مشاهده كرده و جلوی آن را با تيغ اسكالپر تميز علامتگذاری كنيد.
- در جلوی باند يك حفره ايجاد كنيد (عرض حفره مساوي عرض باند باشد).
- يك قطعه كاغذ واتمن و يك قطعه كيسه دياليز (Dialysis tubing) به اندازه حفره ببريد.
- كاغذ واتمن را در جلو حفره و كيسه دياليز را در پشت كاغذ قرار دهيد.
- الكترو فورز را ادامه دهيد (بافر تانك را كمتر كنيد بهطوریکه روي ژل را نپوشاند).
- وقتي DNA وارد كاغذ شد، الكترو فورز را متوقف كنيد.
- كاغذ را جمع كرده و به لوله 0/5 میلیلیتري كه انتهاي آن سوراخ شده است، انتقال دهيد.
- توسط سمپلر مقدار مناسب از بافر زیر روي كاغذ بريزيد: (pH=7.5)
- 150mM NaCl
- 20mM Tris
- 1mM EDTA
- 5%SDS
10- لوله مذكور را داخل يك لوله بزرگتر قرار دهيد (1/5 میلیلیتر)، سانتريفيوژ كنيد تا مايع از ته لوله كوچك به داخل لوله بزرگتر حركت كند، چند دفعه اين كار را تكرار كرده تا موقعي كه ديگر DNA روي كاغذ واتمن مشاهده نشود (با نور UV آزمايش شود).
- اين بافر را جمعآوری و با C.I استخراج كرده و با الكل تغليظ كنيد.
ب) احياي DNA از روي ژل آگارز Low Melting Point (LMP)
1- DNA موردنظر را با آگارز LMP 2% الكتروفورز كنيد و اجازه دهيد تا نوارهاي DNA خوب تفكيك شوند.
2- با نور UV باند موردنظر را مشاهده كنيد.
3- باند موردنظر را با تيغ اسكالپر تميز علامتگذاری كنيد.
4- ژل را روي كاغذ سفيد قرار داده و باند موردنظر را بريده به لوله تمیز دیگر منتقل كنيد.
5- قطعه ژل را يك شب در فريزر 20- درجه قرار دهيد.
6- قطعه ژل را در حرارت اتاق قرار دهيد و سپس 5 دقيقه در آب 65 درجه انكوبه كنيد.
7- محلول حاصل را به دمای اتاق رسانده و دو مرتبه با P.C.I استخراج كنيد تا ژل از آن حذف شود.
- استات سديم (غلظت نهايي 3M) را به محلول اضافه كرده با الكل رسوب دهيد.
استخراج پلاسميد در مقياس كم (Mini preparation)
استخراج پلاسمید با روش آلكالين و بهصورت Mini preparation در موارد غربالگری کلنی[4] استفاده میشود، ولی براي كارهاي حساستر، پلاسميد بهصورت حجم بالا[5] استخراج میگردد. براي استخراج پلاسميد، سلول باكتري با محلول GTE (گلوكز، تريس، EDTA) ليز شده و محتويات آن آزاد میشود. DNA ژنومی و پروتئینهاي سلولي را با محلول قليا (NaOH و SDS) دناتوره كرده (در اين روش DNA سوپركويل دناتوره نمیشود) و بلافاصله با اسيدي كردن محيط و ایجاد سرما DNA ژنومی و پروتئینها را رسوب میدهیم. در اين فرآیند پلاسمید بهصورت محلول باقي میماند که توسط سانتريفيوژ آن را از فاز آلی (پروتئینها) جدا میکنیم.
روش كار:
- مقدار 1/5 میلیلیتر كشت شبانه (باكتري حاوي پلاسميد) را به مدت 5 دقيقه با 4500rpm سانتريفيوژ كنيد.عدهای معتقدند چنانچه رسوب را با محلول STE سوسپانسيون كرده و دوباره سانتريفيوژ كنيم (شستشو دهيم) واکنشهای آنزيمي بهتر انجام میشود.
STE: (pH=8)
- 100mM NaCl
- 10 mMTris
- 1mM EDTA
- رسوب را در 100μl محلول Iبهصورت سوسپانسيون درآورید (محلول روي يخ سرد شده باشد).
محلول I: (pH= 5)
- 8mM glucose
- 25mm Tris
- 10mM EDTA
3-. ميكروتيوب حاوي واكنش را مدت 10-5 دقيقه در هواي اتاق قرار دهيد.
4.- مقدار 200μl از محلول II تازه تهیهشده را به لوله واكنش اضافه كرده، درب لوله را ببنديد و با وارونه كردن آن محلول را خوب مخلوط کنید و 5 دقيقه روي يخ قرار دهيد.
محلول II:
- 2N NaOH
- 1% SDS
5- مقدار 150μl از محلول III (سردشده روي يخ) را به لوله واكنش اضافه كنيد. درب لوله را بسته و به آهستگي 10 دفعه آن را وارونه كنيد و مدت 15 دقيقه روي يخ قرار دهيد.
محلول III:
- 60 میلیلیتر 5M AcoK
- اسيد استيك 11/5 میلیلیتر
- آب مقطر 28/5 میلیلیتر
اين محلول داراي 3 مولار پتاسيم و 5 مولار استات است. وقتي اين محلول به واكنش اضافه میشود بايد يك رسوب سفيد رنگ در داخل لوله پديدار گردد.
6- مدت 20 دقيقه با 12000xg آن را سانتريفيوژ كنيد.
7- محلول رويي (تقریباً 450 ميكروليتر) را به لوله ديگر انتقال دهيد (حاوي پلاسميد میباشد).
8- سديم استات با غلظت نهايي 0.3M را به آن اضافه كنيد (عدهای اين مرحله را ضروری نمیدانند).
9- دو برابر حجم آن اتانل مطلق سرد اضافه كنيد.
10- مدت 15 دقيقه آن را سانتريفيوژ كنيد تا پلاسمید رسوب كند. الكل را حذف كنيد (حتماً لوله را وارونه روي كاغذ جاذبالرطوبه قرار دهيد).
11- روي رسوب الكل 70 درجه (100 ميكرليتر) اضافه كنيد و آن را ورتكس كنيد، بهطوریکه رسوب كنده شده و در الكل شناور شود.
12- مدت 3 دقيقه در حرارت اتاق آن را سانتريفيوژ كنيد (شستشو دادن با الكل).
13- الكل را خالي كرده و رسوب را در حرارت 70 درجه خشککنيد.
14- رسوب را در 50 ميكروليتر آب يا بافر ( TE) حل كنيد.
TE= pH 8
- 10mM Tris
- 1mM EDTA
استخراج پلاسميد در مقياس بالا (Large scale Plasmid DNA preparation):
- مقدار 500 میلیلیتر کشت شبانه را در 4000g به مدت 10 دقيقه سانتريفيوژ كنيد. مايع رويي را خالي كرده و لوله را وارونه روي كاغذ خشککن قرار دهيد تا خشک شود. رسوب را با STE سوسپانسيون نموده و دوباره سانتزيفيوژ كنيد و سپس لوله را وارونه قرار داده تا خشک شود.
STE = (pH=8)
- 100mM NaCl
- 10 mMTris- base
- 1mM EDTA
- رسوب را در 10 میلیلیتر محلول Iحل كنيد.
Solution I: (pH=8)
- 50 mM glucose
- 25 mM Tris
- 10 mM EDTA
- آن را خوب سوسپانسيون كنيد و 10 دقيقه در هواي اتاق قرار دهيد.
- مقدار 20 میلیلیتر از محلول II تازه تهیهشده را به آن اضافه كرده، درب لوله را ببنديد و با وارونه كردن آن محلول را خوب مخلوط كنيد و 5 دقيقه روي يخ قرار دهيد.
Solution II:
- 2N NaOH
- 1% SDS
- 5. مقدار 15 میلیلیتر از محلول III به واكنش اضافه كنيد و 15 دقيقه لوله را روي يخ قرار دهيد.
Solution III:
- 60 ml of 5M ACOK,
- 5 ml of Acetic Acid,
- 5 ml of ddH2O
(اين محلول داراي 3 مولار پتاسيم و 5 مولار استات است). وقتي اين محلول اضافه میشود بايد يك رسوب سفيد رنگ پديدار گردد (میتوان با پر و خالي كردن محلول توسط پيپت، پروتئینها را شكست).
- مدت 20 دقيقه با 20000xg آن را سانتريفيوژ كنيد.
- محلول روييرا (تقریباً مقدار 15 میلیلیتر) به لوله جديد انتقال دهيد.
- مقدار 0/6 حجم اوليه (12 میلیلیتر) ايزوپروپانلرا به آن اضافه كنيد و خوب مخلوط كرده و 15 دقيقه در حرارت اتاق قرار دهيد.
- در حرارت اتاق و مدت 15 دقيقه آن را سانتريفيوژ كنيد تا پلاسمید در ته لوله رسوب كند (توجه: اگر در 4 درجه انجام شود، نمکهارسوب میکنند).
مايع رويي را حذف كنيد (حتماً لوله را وارونه قرار دهيد) و رسوب را با الكل 70 درجه و در حرارت اتاق شستشو دهيد (مقدار 10 میلیلیتر الكل 70 درجه به هر لوله اضافه كرده و با g17000 به مدت 10 دقيقه سانتريفيوژ كنيد). الكل را خالي كرده و آن را در حرارت 70 درجه خشک کنيد. رسوب را با 8 میلیلیتر آب يا بافر TE حل كنيد.
[1] Dimethyl sulfoxide
[2] هیدروکسی اتیل آمینواتیل هیدروکلرید
[3] Phenol/chloroform/isoamyl alcohol
[4] Colony screening
[5] Large scale
استخراج و خالصسازی DNA پلاسمیدی از سودوموناس آئروجینوزا
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام