ژنومیکس و کاربرد آن در تشخیص بیماریها
سینا وکیلی دانشجوی کارشناسی ارشد بیوشیمی بالینی گروه بیوشیمی دانشگاه علوم پزشکی کرمان
دکتر غلامرضا اسدی کرم استاد بیوشیمی بالینی گروه بیوشیمی دانشگاه علوم پزشکی کرمان

ژنومیکس مطالعه کل ژنوم است که شامل مطالعه توالی، نوترکیبی، عملکرد و ساختار میباشد (1). مطالعه یک ژن منفرد زمانی ژنومیکس نام میگیرد که تأثیر آن بر کل ژنوم یا پاسخی که به ژنوم میدهد (تأثیری که از ژنوم میگیرد) مورد بررسی واقع شود (2).


| فریدریک سانگر(بالا) و والتر گیلبرت (پایین) |
فردریک سانگر و همکارانش علاوه بر یافتن توالی آمینواسیدی انسولین نقش کلیدی در گسترش روشهای تعیین توالی DNA را ایفا کردند؛ بدین صورت که در سال 1975 وی و Alan Coulson یک روش تعیین توالی به نام Plus and Minus ابداع کردند که در آن از DNA پلیمراز و همچنین اسیدهای نوکلئیک رادیولیبل شده استفاده میشد (11). این روش قادر به تعیین توالی DNA تا 80 نوکلئوتید بود (11). فردریک سانگر با اعمال تغییراتی در روش Plus and Minus روش Chain termination یا همان روش سانگر را ابداع نمود که اساس تکنیکهای توالی یابی DNA را پایه گذاری کرد (12). در همین زمان Walter Gilbert و Allan Maxam نیز به طور جداگانه روشی با کارایی کمتر نسبت به روش سانگر طراحی کردند که روش شیمیایی یا روش ماکسام و گیلبرت (DNA sequencing by chemical degradation) نام گرفت (13).
سانگر و گیلبرت به خاطر توسعه روشهای توالی یابی DNA نیمی از جایزه نوبل شیمی را در سال 1980 دریافت کردند (14).واژه ژنومیکس برای اولین بار توسط دکتر Tom Roderick که یک ژنتیکدان بود مورد استفاده قرار گرفت (3). به دنبال کشف ساختمانDNA توسط واتسون و کریک و همچنین انتشار توالی آمینواسیدی انسولین توسط Fred sanger اسیدهای نوکلئیک مورد توجه بیولوژیستها قرار گرفتند (4). در سال 1964 اولین توالی اسید نوکلئیک توسط Robert W Holley منتشر شد که این توالی مربوط به tRNA آلانین بود (5, 6). بعد از آن Marshal Nirenberg و Philip Leder ماهیت سهتایی کدهای ژنتیکی (کدونها) را آشکار کردند (7). در سال 1972 Walter Fiers و تیمش برای اولین بار توالی یک ژن را تعیین کردند که ژن مربوط به پروتئین پوششی باکتریوفاژ MS2 بود(8). آنها در ادامه کارشان توالی نوکلئوتیدی کل ژنوم باکتریوفاژ MS2 (که 3569 جفت باز بود) را شناسایی کردند. در ادامه توالی نوکلئوتیدی ژنوم Simian virus 40 نیز در سال 1976شناسایی گردید (9, 10).
اولین توالیهای یوکاریوتی که بعد از ابداع روش فوق تعیین شدند توالی ژنومی میتوکندری و کلروپلاست بودند (15). در ادامه توالیهای بسیاری که اکثراً مربوط به پاتوژنهای مختلف بیماریزا بودند تعیین گردید (16). مطالعه و تعیین توالی کل ژنوم انسان با عنوان Human Genome Project در سال 2003 با تعیین توالی کل ژنوم یک فرد به پایان رسید و در سال 2007 این توالی با کمتر از یک خطا در 20000 باز به ثبت رسید (17). با توسعه تکنولوژیهای توالییابی، پروژههای ژنومی زیادی به ثبت رسید که ادامه این روند با محدودیتهای سیاسی و اجتماعی روبرو میباشد (18).
 Francis_Crick
Francis_Crick

James_D_Watson
 Walter_Fier
Walter_Fier

Robert_W._Holley
تصاویر تعدادی از افرادی که در پیشرفت علوم ژنتیک و ژنومیکس نقش مهمی ایفا کردند
کاربردها و ابعاد مختلف ژنومیکس
ژنومیکس را میتوان از زوایای مختلفی مورد بررسی قرار داد:
Functional Genomics:
در ژنومیکس عملکردی، تفسیر، عملکرد و تعامل بین ژنها با استفاده از اطلاعات ژنومی مورد بررسی قرار میگیرد. Functional genomics در واقع به بررسی عملکرد DNA در سطح ژن، رونویسی RNA و محصولات پروتئینی میپردازد و هدف اصلی آن یافتن ارتباط بین ژنوم یک ارگانیسم با فنوتیپ آن میباشد (19).
Structural Genomics:
ژنومیکس ساختاری، ساختمان سه بعدی تمام پروتئینهایی که توسط ژنوم کد میشوند را تشریح میکند. با توجه به ارتباط نزدیک ساختمان و عملکرد پروتئینها، ژنومیکس ساختاری پتانسیل توضیح عملکرد پروتئینها را دارد (20). یکی از اهداف ژنومیکس ساختاری شناسایی ساختمانهای جدید پروتئینی است (21).
Epigenomics:
مطالعه تمام ویژگیهای اپیژنتیکی ژنوم را اپیژنومیکس میگویند که از جمله مهمترین این ویژگیها متیلاسیون DNA و Histone modification میباشد. اپیژنتیک نقش مهمی در تنظیم بیان ژن ایفا میکند (22).
Metagenomics:
متاژنومیکس مطالعه متاژنوم میباشد. متاژنوم محصولات ژنتیکی هستند که مستقیماً از نمونههای موجود در محیط به دست میآیند. متاژنومیکس را میتوان به صورت زیر تعریف کرد:
” بکارگیری تکنیکهای مدرن ژنومیکس در جوامع میکروبی، مستقیماً در محیط طبیعی آنها بدون نیاز به ایزوله کردن یا کشت گونههای خاص در آزمایشگاه” (23).
تکنیکها، روشها و ابزارهای مورد استفاده در ژنومیکس
ابزارها و روشهای مختلفی در ژنومیکس مورد استفاده قرار میگیرند که در ادامه به اختصار توضیح داده میشوند.

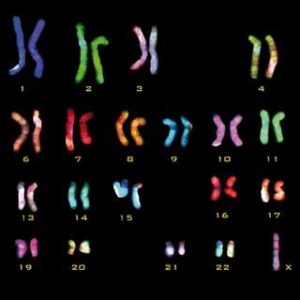
شکل 1. تصویر یک کاریوگرام معمولی به همراه دسته بندی کروموزومها
کاریوتایپ بررسی کروموزومهای یک سلول است که در مرحله متافاز متـــــــــوقف شده و با یک رنگ خاص رنگآمیزی و قابل رؤیت گردیدهاند (شکل 1). با پیشرفت تکنیکهای tissue culture و بکارگیری روشهای رنگآمیزی کروموزومها، برای اولین بار کاریوگرامها در سالهای بین 1950 تا 1960 ارائه شدند (24). کاریوگرامها نشان دهنده اندازه، شکل و محل قرار گیری کروموزومها میباشند (24).
برای انجام کاریوتایپ بر روی انسان، معمولا از گلبولهای سفید خونی که به راحتی کشت میشوند استفاده میگردد (25). این سلولها در طی تقسیم سلولی و در مرحله میتوز به وسیله محلولی از colchicine متوقف شده و کروموزومها آزاد میگردند (26). برای اینکه کروموزومها قابل رؤیت گردند از تکنیکهای مختلفی استفاده میشود؛ یکی از این تکنیکها G-banding یاGiemsa-banding است. در این تکنیک کروموزومهایی که در مرحله متافاز متوقف شدهاند در معرض تریپسین قرار گرفته و سپس به وسیله رنگ گیمسا رنگآمیزی میگردند، سپس به وسیله میکروسکوپ از آنها عکسبرداری شده و نتایج به صورت کاریوگرام مرتب میشوند. نواحی غنی از A و T بصورت باندهای تیره مشخص میگردند (26, 27). روش دیگر banding نیز وجود دارد از جمله آنها میتوانR-banding (رنگآمیزی نواحی تیره یوکروماتین)، C-banding (سانترومرها)، Q-banding (استفاده از quinacrine و بررسی الگوی فلورسنت) و T-banding (تلومرها) را نام برد (25).
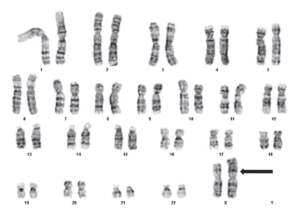
|
شکل 2. تصویر کاریوگرام دختری با سندرم ترنر. ایزوکروموزوم X با پیکان مشخص شده است |
اگرچه تکنیکها و تکنولوژیهای ژنومیکس پیشرفتهای زیادی داشتهاند، اما روشهایی مانند کاریوتایپینگ امروزه نیز برای اهداف غربالگری و یا در نواحی دارای امکانات محدود همچنان مورد استفاده قرار میگیرند. از کاریوتایپینگ برای تشخیص بیماریهای ژنتیکی مختلفی استفاده میشود (شکل 2). از جمله این بیماریها میتوان به سندرم داون (شکل 3) و سندرم ترنر (Turnuer) اشاره کرد (24).

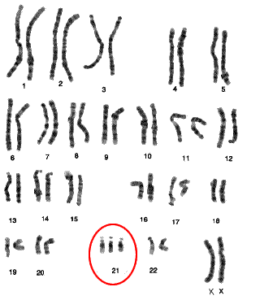
شکل 3. (سمت چپ) کاریوگرام مربوط به فردی با سندرم داون که تریزومی 21 در آن مشخص شده است. (سمت راست) مشخصات چهره افراد دارای سندرم داون
Fluorescence in situ hybridization (FISH):
|
شکل 4. (سمت چپ) نتیجه حاصل از هیبریداسیون پروبهای FISH با کروموزومهای متافاز. (سمت راست) یک پروب نشاندار که به توالی هدف خود متصل شده است |

در دهه 1960، Joseph Gall و Mary Lou Pardue دریافتند که برای یافتن محل توالیهای DNA در جایگاه طبیعی خود در کروموزوم میتوان از هیبریداسیون مولکولی استفاده کرد (شکل 4). in situ مفهوم “در جایگاه طبیعی آنها در کروموزوم” را میرساند. در ابتدا فرآیند نشاندار کردن به وسیله مواد رادیواکتیو انجام میگرفت اما اندکی بعد لیبلهای فلورسنت جایگزین آنها شدند (28).
اولین مرحله در FISH تهیه یک پروب نشاندار فلورسانس و یا یک پروب تغییر یافته است که بتوان پس از اتصال آن را نشاندار کرد. ابتداDNA هدف و پروب بوسیله حرارت یا بصورت شیمیایی دناتوره میشوند و سپس با یکدیگر مخلوط میگردند. پروب به قسمتی از DNA که مکمل آن میباشد متصل میگردد. محل قرار گرفتن توالی هدف بر روی کروموزوم بوسیله میکروسکوپ فلورسانس مشخص میشود. امروزه این تکنیک به سمت تشخیص بالینی سوق پیدا کرده است. از این تکنیک میتوان به منظور تشخیص اختلالات کروموزومی نظیر حذف شدن قطعه خاصی از DNA، دریافت نسخه اضافی و جابجاییهای کروموزومی استفاده کرد (28).
Multifluor FISH (Spectral karyotyping):
شکل 5. نتیجه حاصل از Multifluor FISH که در آن کروموزومها با رنگهای مختلف مشخص شدهاند |
برای تشخیص بازآرایی های کروموزومی و بررسی سریع مجموعه کروموزومها میتوان از این تکنیک استفاده کرد. نتیجه حاصل از FISH Multifluor در واقع کاریوتیپی است که در آن کروموزومها با رنگهای متفاوتی رنگآمیزی شدهاند (شکل 5). هر رنگ در واقع مجموعهای از پروبهاست که به نواحی مختلف کروموزوم اتصال یافتهاند. یک کروموزوم طبیعی در تمام طول خود به یک رنگ دیده میشود اما کروموزوم های غیر طبیعی مانند کروموزومهای سلولهای سرطانی، ظاهری راه راه با رنگهای دیگر را نشان میدهند (28).
این روش نیز مانند روش کاریوتیپ دارای قدرت تفکیک پایینی نسبت به سایر روشها میباشد و حذف یا اضافه شدن قطعات DNA با اندازه کوچک در آنها قابل تشخیص نیست. برتری این روش نسبت به کاریوتیپ، توانایی بررسی کروموزومهای اینترفازی است و نیازی به کشت سلولها به مدت طولانی نیست، همچنین در این روش میتوان نواحی مختلفی از کروموزوم را به طور همزمان مورد بررسی قرار داد (28).

شکل 6. قسمتی از نتیجه یک Microarray. هر نقطه نشان دهنده یک الیگونوکلئوتید خاص است که با پروب نشاندار مربوط به خود هیبرید شده است |
ایده و متدولوژی Microarray برای اولین بار بوسیله Antibody microarrayها معرفی گردید (29). DNA Microarrayها شامل پروبهای DNA میباشند که به یک سطح جامد مانند شیشه یا سیلیکون متصل شدهاند. هر نقطه بر روی این سطح جامد دارای تعداد زیادی پروب است که قطعات DNA میتوانند به آنها متصل شوند. قطعات DNA که با یک ماده فلورسانس نشاندار شدهاند به قطعه پروبی که مکمل آنها است متصل میگردند و سپس نقاطی که قطعات DNA به آنها متصل شده و یا متصل نشدهاند مورد بررسی قرار میگیرند (شکل 6). این تکنیک کاملاً مشابه تکنیکهای قدیمی ساترن بلات و نورترن بلات بوده و تفاوتش با این روشها در این است که در Microarray تعداد زیادی ژن بطور همزمان مورد بررسی قرار میگیرند (30).
با بکار گیری این تکنولوژی میتوان میزان بیان هزاران ژن را به طور همزمان مورد بررسی قرار داد (30). از این تکنولوژی میتوان در تشخیص و افتراق تعداد زیادی از سرطانها مانند سرطان پروستات و سرطان سینه استفاده کرد؛ بعنوان مثال برای افتراق لوسمی میلوبلاستی حاد ( AML) و لوسمی لنفوبلاستی حاد (ALL) از این تکنولوژی استفاده شده است؛ بدین صورت که مجموعهای 38 تایی از نمونههای لوسمی جمعآوری و بیان 6817 ژن انسانی در آنها مورد بررسی قرار گرفت 50 ژن به صورت یک زیر مجموعه از این مجموعه ژنی برای افتراق AML و ALL مورد استفاده قرار گرفت که از جمله آنها میتوان CD-11، CD-33 و MB-1 که پروتئینهای سطحی سلول را کد میکنند را نام برد. ژن دیگر گیرنده لپتین بود که مارکری در لوسمی حاد محسوب میشود و در تنظیم وزن نقش دارد و مشخص شد که در AML دارای افزایش بیان میباشد. ژنهای مربوط به چرخه سلولی، آپوپتوز و سایر انکوژنها نیز مورد بررسی قرار گرفتند (30, 31).
از DNA Microarray میتوان در شناسایی یک مسیر متابولیکی خاص، بیماری خاص و یا برای اهداف درمانی استفاده کرد؛ به عنوان مثال کاتپسین k که یک سیستئین پروتئاز میباشد به طور انتخابی در استئوکلاستها بیان میشود. استئوکلاستها مسئول بازجذب استخوانها هستند. عدم تعادل بین تشکیل استخوان و بازجذب آن میتواند منجر به استئوپورز شود. با بررسیهای انجام شده مشخص شد کاتپسین k به طور انتخابی در استئوکلاستها بیان میگردد و مهار کردن آن میتواند باعث جلوگیری از استئوپورز گردد (32).
از Microarray میتوان در تشخیص و شناسایی پاتوژنها نیز استفاده کرد. با استفاده از این تکنولوژی میتوان باکتری پاتوژن Escherichia coli o.157:hv را از باکتری غیر پاتوژن E.coli K12 افتراق داد (30). بنابراین از آن جایی که تکنولوژی Microarray امکان بررسی تعداد زیادی از ژنها و میزان بیان آنها را به طور همزمان فراهم میکند، میتوان از آن به عنوان ابزار مناسبی برای اهداف تشخیصی در شرایط مختلف استفاده کرد (30).
Array comparative genomic hybridization (aCGH):
زمانی که اصطلاح Array comparative genomic hybridization برای اولین بار مورد استفاده قرار گرفت، این تکنیک بیشتر به تکنیک FISH شبیه بود و شباهت کمتری به تکنولوژی Array داشت (33, 34). aCGH در واقع ترکیبی از اصول تکنولوژی CGH (شکل 7) و Array میباشد. برخلاف روش CGH که در آن از کروموزومهای متافاز استفاده میشد، در aCGH نیازی به کروموزومهای متافازی نیست و آنالیز کروموزوم با استفاده از قطعات DNA امکان پذیر است. پروبهای DNA با توالی مشخص بر روی یک ماتریکس جامد مانند شیشه فیکس میشوند. به دلیل اینکه توالی پروبهای استفاده شده کوتاه است، قدرت تفکیک aCGH از روشهای CGH قدیمی بسیار بیشتر میباشد. قدرت تفکیک به طول پروبها و فاصله ژنومی بین آنها بستگی
دارد (35).

شکل 7. روش CGH
در aCGH ابتدا DNA نمونه استخراج میگردد و به وسیله یک رنگ فلورسانس معین نشاندار میشود. یک نمونه از DNA نرمال نیز به عنوان کنترل با رنگ فلورسانس دیگری نشاندار میشود. DNAهای تست و کنترل با یکدیگر مخلوط شده و بر روی آن Microarray انجام میگیرد. از آن جایی که قطعات DNA دناتوره و بصورت تک رشتهای میباشند، به قطعات پروب مکمل خود بر روی سطح جامد متصل میگردند، سپس میزان سیگنال فلورسانس تست نسبت به کنترل اندازهگیری میشود. اگر سیگنال فلورسانس نمونه بیشتر بود، نشان دهنده دریافت این قطعه ژنی توسط ژنوم میباشد و برعکس، اگر سیگنال فلورسانس کنترل در یک ناحیه از ژن بیشتر از سیگنال فلورسانس مربوط به تست بود، نشان دهنده این است که این قسمت از ژنوم در نمونه حذف گردیده است. آن نواحی ژنومی که در بین تست و کنترل یکسان هستند، دارای سیگنالی با نسبت مساوی از هر دو نوع فلورسانس لیبل شده میباشد. ازaCGH میتوان در تشخیص بازآراییهای کروموزومی و همچنین حذف قطعاتDNA با کارایی بالاتر نسبت به تکنیک FISH استفاده کرد (35, 36).
|
شکل 8. روش aCGH |

Multiplex ligation-dependent probe amplification (MLPA):
MLPA یک روش Multiplex PCR میباشد که قادر است تعداد کپیهای غیرطبیعی موجود در DNA یا RNA را شناسایی کند (37). در روش MLPA توالی هدف تکثیر نمیشود، بلکه پروبهای MLPA که به توالی هدف متصل شدهاند تکثیر میشوند. برخلاف Multiplex PCR، در این روش فقط از یک جفت پرایمر استفاده میشود.
MLPA دارای 5 مرحله است:
- دناتوره کردن DNA هدف و هیبرید شدن پروبهای MLPA به آن
- واکنش اتصال (ligation)
- واکنش PCR
- جداسازی محصولات PCR به وسیله الکتروفورز
- آنالیز دادهها
در مرحله اول DNA دناتوره میشود و بصورت Overnight با مخلوطی از پروبهای MLPA انکوبه میگردد. پروبهای MLPA شامل دو الیگونوکلئوتید مجزا میباشد که هر کدام از آنها دارای یکی از پرایمرهای PCR است (در واقع قطعه پروبی که مکمل توالی هدف میباشد از دو قسمت تشکیل شده است

شکل 9. روش MLPA |
که پس از اتصال به ناحیه هدف در کنار یکدیگر قرار میگیرند). فقط زمانی که هر دو الیگونوکلئوتید مربوط به یک قسمت از توالی هدف متصل شدند واکنش Ligation اتفاق میافتد و دو الیگونوکلئوتید به یکدیگر متصل میگردند. پس از اتصال دو الیگونوکلئوتید، بر روی این پروب (که هماکنون با اتصال دو قسمت خود کامل شده است) PCR انجام میگیرد. با توجه به اینکه توالی مربوط به پرایمر همه پروبها یکسان میباشد، برای PCR فقط از یک جفت پرایمر استفاده میشود (شکل 9). همچنین در این روش نیازی به شستشوی پروبهای متصل نشده نیست چون پروبهای متصل نشده نمیتوانندPCR شوند (38).
از آن جایی که هر پروب توالی منحصر به فردی دارد، محصولات حاصل از PCR بوسیله الکتروفورز (مویینه) جدا میگردند. پرایمر Forward با یک رنگ فلورسانس نشاندار است، بنابراین هر قطعه یک peak فلورسانس ایجاد میکند که به وسیله capillary sequencer قابل ردیابی میباشد. با مقایسه peak مربوط به نمونه و الگوی نمونههای مرجع مقدار هر قطعه مشخص میگردد. این نسبت به دست آمده نشان دهنده میزان توالی هدف موجود در نمونه DNA میباشد (38).
ازMLPA میتوان در تشخیص CVNها (Copy number variations)، جهشهای موزاییک، حالات متیلاسیون و تأیید ناهنجاریهایی که به وسیله FISH یا CGH تشخیص داده نمیشوند استفاده کرد. MLPA نمیتواند بازآراییهای ژنومی متعادل (Balanced genome rearrangements) مانند ترانسلوکاسیونها و Inversionها را تشخیص دهد (39).
پروژههای ژنومی از سه بخش تشکیل میشوند:
- توالی یابی (sequencing)
- مونتاژ توالیهای به دست آمده برای یافتن توالی اصلی (assembling)
- حاشیه نویسی (annotation) و آنالیز توالی بدست آمده (40)
با پیشرفت تکنولوژیهایی که قادر هستند تمامی ژنوم را مورد بررسی قرار دهند احتمالاً توالییابی ژنوم در آینده بعنوان اولین ابزار آنالیز ژنتیکی مورد استفاده قرار گیرد (24).
توالییابی:
بطور کلی روشهای توالییابی ژنوم shotgun و high-throughput(next generation sequencing) میباشند (40).
در واقع همان روش End termination سانگر میباشد و میتوان آن را برای قطعات بالای 1000 جفت بازی به کار برد (41). از آن جایی که روش End termination برای قطعات 100 تا 1000 جفت بازی به کار میرود قطعات بزرگتر DNA به صورت تصادفی به قطعات کوچکتر شکسته میشوند و این قطعات کوچک توالییابی میشود، سپس به وسیله نرم افزارهای کامپیوتری همپوشانی انتهای رشتههایی که توالییابی شدهاند بررسی میگردد (شکل 10) و توالی این قطعات بصورت یک رشته پیوسته گردآوری میگردد (41, 42). استفاده از توالییابی سانگر به تنهایی برای تشخیص بسیاری از اختلالات ژنتیکی مناسب نیست (24).

شکل 10. توالی یابی DNA به روشShotgun . علت نامگذاری این روش به نام Shotgun یا تفنگ ساچمهای، قطعه قطعه شدن تصادفی DNA میباشد که به الگوی تصادفی ساچمهها در تفنگ ساچمهای مربوط است |
نسلهای جدید توالییابی در جهت کاهش هزینهها و افزایش سرعت توسعه یافتهاند. یکی از این روشهاIllumina ( Solexa ) sequencing میباشد (43).
این روش بر پایه خاتمه رنگی برگشت پذیر (Reversible dye-termination) استوار میباشد که به ما این امکان را میدهد تا تک تک بازها را در هنگام سنتز DNA شناسایی کنیم. مولکول DNA قطعه قطعه شده بوسیله پرایمرهایی که به یک سطح جامد متصل شدهاند تکثیر شده و کلنیهایی از قطعات مختلف بصورت متصل به صفحه جامد ایجاد میگردد، پس از این مرحله نوکلئوتیدهایA ,T ,C و G که انتهای ‘3 آنها بلاک شده و با رنگهای مختلف نشاندار شدهاند، اضافه میشوند. نوکلئوتیدی که مکمل اولین باز است متصل میگردد و سایر نوکلئوتیدها شسته میشوند. پس از هر بار سنتز، پرتوهای لیزر تابانده میشوند و گروه بلاک کننده آزاد شده و رنگ فلورسانس مخصوص هر باز قابل تشخیص میشود (شکل 11). این رنگ ثبت میگردد و چرخه بعدی با اضافه کردن مجدد نوکلئوتیدها آغاز میگردد (44).
تکنولوژی توالییابی DNA نمیتواند کل ژنوم را بصورت پیوسته توالییابی کند. Assembling در واقع ردیف کردن و یکی کردن قطعات توالییابی شده برای یافتن توالی اصلی DNA میباشد (40). فرآیند Assembling به دو دسته تقسیم میشود:
De novo: برای ژنومهایی که مشابه هیچکدام از توالیهایی که در گذشته شناسایی شدهاند، نمیباشد.
Comparative: توالی هدف مشابه توالیهایی است که قبلاً تعیین شدهاند (45). پس از انجام این مرحله، توالی یک رشته پیوسته بدست میآید (46).

شکل 11. توالی یابی DNA به روش Illumina (Solexa) |

تفسیر ژنوم فرآیندی است که در چند گام اساسی اطلاعات بیولوژیکی ژنومی که توالییابی شده را فراهم مینماید (47). این گامها شامل موارد زیر میباشند:
- مشخص کردن قسمتهایی از ژنوم که پروتئینها را کد نمیکنند.
- Gene prediction که مشخص کردن قسمتهایی است که ژنها را کد میکنند.
- افزودن اطلاعات بیولوژیکی قسمتهای کد کننده ژن (48).
تفسیر ژنوم توالییابی شده به دلیل گزارشها و نتایج کمی که تاکنون از آن وجود دارد محدود و مشکل میباشد (24).
جمعبندی و مقایسه روشها و تکنیکهای مورد استفاده در ژنومیکس
اگرچه امروزه با پیشرفت تکنولوژی، امکان بررسی تمام ژنوم بصورت نوکلئوتید به نوکلئوتید با استفاده از روشهای Whole genome sequencing (WGS) و Whole exome sequencing (WES) با سرعت بیشتر و هزینه کمتر نسبت به قبل وجود دارد، اما همچنان تکنولوژیهای مولکولی Non sequencing به طور گسترده و برای اهدافی مانند غربالگری و یا در مناطقی که دسترسی به امکانات پیشرفته وجود ندارد مورد استفاده قرار میگیرند (24). تستهای ژنومیکس در محدوده وسیعی از جمله غربالگری نوزادان، تستهای تشخیصی برای اختلالات ارثی، تستهای پیشبینی کننده برای اختلالات ناگهانی و شدید بزرگسالان و تستهای فارماکوژنتیکی برای انتخاب دارو و دوز آن مورد استفاده قرار میگیرند (24).
همانطور که گفته شد، برای بررسیهای ژنومیکس از تستها و تکنیکهای مختلفی استفاده میشود که آنها را میتوان از جهات مختلف مقایسه کرد. تستهای ژنتیک بالینی که بیشترین کاربرد را دارند در جدول 1 آورده شدهاند. حساسیت، ویژگی، هزینه، مدت زمان انجام تست و قابلیتهای یک تست از جمله مواردی هستند که مورد توجه قرار گرفته اند(24).

جدول 1. تستهای ژنتیکی بالینی که بیشترین کاربرد را دارند
هر گونه تغییر و نوآوری در تکنولوژیهای تشخیصی نیاز به ارزیابی کیفیت و سهولت آن دارد. از جمله اصطلاحاتی که برای ارزیابی تستهای تشخیصی مورد استفاده قرار میگیرند Analytical validity و Clinical validity میباشند که به صورت زیر تعریف میشوند:
Analytical validity: توانایی یک تست مولکولی در تشخیص واریانتهای ژنتیکی، هم از نظر حساسیت (میزان منفی کاذب) و هم از نظر ویژگی (میزان مثبت کاذب).
Clinical validity: توانایی یک تست در پیشبینی وجود یا عدم وجود یک حالت بالینی خاص (24).
در جدول 2 اصطلاحاتی که در ارزیابی یک تست تشخیصی مفید میباشند آورده شده است.

جدول 2. اصطلاحات مفید در ارزیابی یک تست ژنتیکی |
تستهای ژنتیکی به دو دسته مســـــــــــــــتقیم ( (Direct genetic testingو غیرمستقیم (Indirect genetic testing) تقسیمبندی میشوند. در تست مستقیم، آزمایشگاه به دنبال واریانتهای ژنتیکی که مستقیماً با بیماری مرتبط هستند، میباشد، اما تست غیرمستقیم به بررسی آن دسته از مارکرهای DNA میپردازد که با شرایط ایجاد شده ارتباط دارند، اما دلیل اصلی ایجاد آن حالت ژنتیکی نمیباشند (24).
در انتخاب یک تست ژنتیکی شرایط فردی از جمله سن بیمار، سابقه خانوادگی و موارد دیگری مورد توجه قرار میگیرند. جدول 3 فاکتورهایی که در انتخاب یک تست ژنتیکی مؤثر هستند را توضیح میدهد (24).
تقسیم بندی واریانتهای ژنتیکی حاصل از تستهای تشخیصی
در تستهای تشخیصی ژنتیک بالینی واریانتها را در یکی از این 6 گروه دسته بندی میکنند:
- Disease causing: تغییر توالی که قبلاً گزارش شده و مشخص شده که علت ایجاد یک اختلال میباشد ( مانند حذف F508 در CFTR) .
- Likely disease causing: تغییر توالی که قبلاً گزارش نشده است اما از انواعی است که انتظار میرود مسؤل ایجاد یک اختلال باشد (مانند جهش خاموش در یک ژن که انواع مشابه این جهش قبلاً گزارش شده است).
- Possibly disease causing: تغییر توالی که قبلاً گزارش نشده و ممکن است باعث ایجاد یک اختلال باشد و یا نباشد.
- Likely not disease causing: تغییر توالی که قبلاً گزارش نشده و احتمالاً اختلالی نیز ایجاد نمیکند.
- Not disease causing: تغییر توالی که قبلاً گزارش شده و مشخص شده که یک واریانت معمول میباشد.
- Variant of unknown clinical significance: تغییر توالی که مشخص نشده و یا انتظار نمیرود که علت ایجاد یک اختلال باشد، اما همراه با یک تظاهر بالینی مشاهده شده است (24).
جدول 3. فاکتورهای مؤثر در انتخاب یک تست ژنتیکی

بیشتر تغییرات فوق میتواند بر اساس فراوانی در جمعیت، یافتههای بالینی، پایگاههای داده مربوط به جهشها و سایر عوامل مورد تفسیر قرار گیرند (24).
کاربرد ژنومیکس در تشخیص و بررسی بیماریها
تکنولوژی ژنومیکس برای بررسی بسیاری از بیماریها بکار گرفته شده است که در ادامه بطور مختصر کاربرد و نتایج حاصل از آن آورده شده است.
کاربرد ژنومیکس در تعدادی از سرطانها
تكميل پروژه ژنوم انساني سبب جهشي در استفاده از فنآوريهاي ژنومي و پروتئومي براي شناسايي ماركرها به منظور تشخيص زود هنگام سرطان با هدف مولكولي شده است. تعداد ژنهاي انساني شناخته شده و تواليهاي بيان شونده همچنان در حال رشد است و ابزارهاي جديدي براي تحليل اين دادهها بوجود آمده است. تفاوت در بيان ژنها در بافتهاي سالم و بدخيم امكان شناسايي ژنها و مسيرهايي كه در سرطانهاي انساني تغيير ميكنند را فراهم ميكند (49-51).
فنآوريهاي متعددی درجهت كشف ماركرهاي سرطاني، دسته بندي و براي كاربردهاي كلينيكي بكار گرفته شده است. برش ميكروسكوپي توسط ليزر از اولين گروه اين فنآوريها ميباشد. اين روش، جداسازي دقيق سلولهاي توموري، استرومايي و سلولهاي سالم از يك نمونه بيوپسي را ممكن كرده است (52). بررسي بهينه نمونههاي جدا شده ميكروسكوپي امكان تفكيك دقيق رخدادهاي داخل و بين هر كدام از اين زيرواحدهاي بافتي را ميدهد. استفاده از اين روشها در نمونههاي بيماران امكان تشريح تغييرات ژنومي، رويدادهاي بيان ژن و تفاوت بيان ژن، فعال سازي و علامت گذاري پروتئينهاي مختلف در نمونههاي توموري را فراهم كرده است (53, 54). سپس بررسي آنها توسط ابزارهاي قدرتمند و جديد بيوانفورماتيك جهت طبقه بندي بيماران سرطاني و غير سرطاني در گروههاي مربوطه، صورت ميگيرد (55).
توسعه و كاربرد اين فنآوريها در نمونههاي كلينيكي، نويد پيشرفتهايي در زمينه تشخيص زود هنگام سرطان، پيشگیري، و درمانهاي اختصاصي تومور را ايجاد كرده است. با افزايش درك محققين در زمينههاي تغييرات اختصاصي در بيان ژنها و مسيرهاي سيگنال پروتئينها امكان تغيير اساس درمان از روش هيستوپاتولوژيكي به سمت درمان اختصاصي مولكولهاي از تنظيم خارج شده ناشي از برهمكنش تومور ميزبان وجود دارد. البته نميتوان اظهار كرد كه اين روشها به طور كامل جايگزيني براي روشهاي مرسوم امروزي در درمان بيماريها ميباشند. پيشرفتهاي ژنوميك و پروتئوميك در هدايت محققين در انتخاب بهترين روش درمان براي هر بيمار به صورت اختصاصي كمك خواهند كرد (56).
تمامي تكنيكهای ژنومیکس در شناسايي اهداف ناشناخته در ژنوم و پروتئوم تومور نقش دارند. شناسايي تغييرات اختصاصي در DNA ,RNA و پروتئينهاي تومور مستلزم دانستن اتفاقاتي است كه در داخل و اطراف تومور رخ ميدهد. در گذشته توانايي محققين درتعيين جايگاه دقيق اين تغييرات به دليل ضعف در جداسازي انواع خاص سلولي از نمونه پاتولوژي دچار اختلال بود. Cell-scraping، خالص سازي با ستون ميل تركيبي (57)،كشت سلولهاي ناميرا در محيط كشت (58)، كشت همزمان سلولي (59) و تشريح دستي بافتها (60) همگي از روشهاي مورد استفادهاي هستند كه هر يك فوايد و ايرادات خاص خود را دارند.
با وجود اينكه اين تكنيكها سبب پيشرفت پژوهشگران در تهيه ردههاي سلولي خالص براي ارزيابي جريانات داخل سلول شدهاند، اما بخشي از اطلاعات كه تعيين كننده فنوتيپ يك نوع تومور خاص ميشوند از دست خواهد رفت؛ مثلاً محيط اطراف تومور سرطاني نه تنها شامل اجزاء بدخيم اپيتليالي است، بلكه استروماي در برگيرنده و بافت سالم اطراف را نيز شامل ميشود. اين اجزاء ميكروسكوپي با استفاده از گيرندهها، اتصالات سلولي، مولكولهاي سيگنال داخل و بين سلولي به سلولهاي توموري امكان برقراري ارتباط با محيط اطراف را ميدهند و نيز نقش فعالي دركنترل و يا پيشرفت آنها دارند (61).
حذف بخشي از اين اجزا جهت كشت سلولها در محيط كشت آزمايشگاهي برهمكنشهاي سلولي- سلولي و سلولي- ماتريكسي، كه احتمالاً بر رفتار تومور اثرگذار هستند، را مختل مينمايند و در نتيجه تصور غلطي از ساختار و فيزيولوژي تومور در محيط سلول زنده به محقق القا ميكنند (61).
با استفاده از تکنولوژی ژنومیکس امكان ارزيابي هزاران ژن به صورت همزمان توسط تكثير RNA با استفاده از علائم فلورسنت و سپس كاربرد اين رونوشتها بر روي صفحات array كه حاوي مقادير زياد اليگونوكلئوتيد و يا cDNA هستند، وجود دارد (62, 63). وجود ماركر فلورسانس بيانگر حضور و اندازه يك رونوشت cDNA خالص در بين جمعيت مورد مطالعه ميباشد. تفاوت در بيان ژنها ثبت ميشود و الگوهاي بيان ژن با كاربرد همزمان چند Microarray و توسط نرمافزارهاي بيوانفورماتيك مقايسهاي محاسبه ميشود (64, 65).
اطلاعات به دست آمده از بررسي ژنوم تومور ورونوشتهاي آن منجر به پيشرفت در كشف ژنهاي جديد و روشهاي شناسايي سرطان ميشود. يكي از تكنولوژيهاي جديدي كه به تازگي و در سالهاي اخير به منظور تسهيل بررسي ژنوم و پروتئوم سلولهاي سرطاني در دسترس قرار گرفته است، Microarrayهاي بافتي ميباشند. Microarrayهاي بافتي در اصل ابزار مفيدي براي آناليز سريع و مناسب تعداد زيادي از بافتهاي پارافين اندود شده، متعلق به انواع تومورهاست. كاربرد اين تكنولوژي امكان استفاده از پروب يكساني را فراهم ميكند و معيار تفسير نتايج را به استاندارد نزديكتر ميسازد. اين ابزار آناليز بيان چندين ژن و تكثير آنها در يك تومور و يا يك ناحيه از تومور توسط يك روش استاندارد راتسهيل ميكند (66).
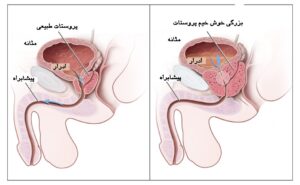
غده پروستات یکی از غدد سیستم تولید مثلی مرد میباشد (شکل 12). در کشورهای توسعه یافته سرطان پروستات شایعترین سرطان غیر پوستی میباشد و عوامل بسیار زیادی از جمله سن، نژاد و سابقه خانوادگی در بروز آن مؤثر هستند. اغلب سرطانهای پروستات دارای رشد کم میباشند اما موارد تهاجمی آن نیز دیده میشود (67). سلولهای سرطانی پروستات ممکن است متاستاز داده و به سایر قسمتهای بدن از جمله استخوانها و غدد لنفاوی منتقل شوند. سرطان پروستات میتواند منجر به درد به هنگام دفع ادرار، مشکلات جنسی و حتی مرگ شود. معمولاً در سنین بالای 50 سال بروز پیدا میکند و ششمین عامل مرگ و میر وابسته به سرطان جهان در مردان میباشد. در کشورهای توسعه یافته شایعترین سرطان بوده و شیوع آن در کشورهای در حال توسعه رو به افزایش است (68, 69).
اخیراً مطالعات ژنومیکس سرطان پروستات به روشن شدن اساس ژنتیکی این بیماری رایج اما پیچیده کمک فراوانی کرده است. مطالعات گسترده ژنومیکس واریانتهای ژنی بیشمار مرتبط با بیماری و همچنین ویژگیهای بیان ژنی در تومورهای پروستات را آشکار کرده است (شکل 13). بر اساس این نتایج، غربالگری فردی ژنی سرطان پروستات میتواند در تشخیص شدت بیماری ارزشمند باشد (70).
شکل 12. غده پروستات
یکی از نتایج حاصل از مطالعات وجود پلیمورفیسم تک نوکلئوتیدی (SNP) در سه ناحیه از کروموزوم 24q8 در مبتلایان به سرطان پروستات بوده است (71, 72). بررسیها نشان دادهاند که این SNPها منجر به افزایش 25 تا 50 درصدی خطر ابتلا به سرطان پروستات میشوند (73). پلیمورفیسمهای تک نوکلئوتیدی ناحیه 24q8 همچنین با سرطانهای سینه، تخمدان، کلورکتال و مثانه مرتبط میباشند (74, 75).
علاوه بر مورد فوق، دو ناحیه مجزا در کروموزوم 17 نیز مشخص گردیده که حدوداً 20 درصد خطر ابتلا به سرطان پروستات را افزایش میدهند (70).
همچنین دو ناحیه مرتبط با سرطان پروستات نیز در کروموزوم 10 مشخص گردیده که یکی از آنها ارتباط پلیمورفیسم (SNP) پروموتر پروکسیمال ژن β-microseminoprotein (MSMB) و این سرطان را نشان میدهد (76, 77). ژن MSMB پروتئین PSP94 (که به نام MSP نیز شناخته میشود) را کد میکند که عضوی از خانواده فاکتورهای متصل شونده به ایمونوگلوبولین میباشد که محرک آپوپتوز و کاهنده رگزایی تومور میباشد (78). پروتئین PSP94 بوسیله سلولهای اپیتلیال پروستات ساخته میشود و یکی از فراوانترین پروتئینهای پروستات و مایع منی است (76). این پلیمورفیسم تک نوکلئوتیدی ژن MSMB تنها دو جفت باز بالاتر از ناحیه شروع رونویسی قرار گرفته و به نظر میرسد که این آلل خطر آفرین، بیان ژن MSMB را در حدود 70 درصد کاهش میدهد (79)، که این یافته همسو با دیگر یافتههایی است که نشان دادهاند کاهش بیان MSMB درگیری سرطان پروستات و پیشرفت آن را افزایش میدهد (78).
جدول 4. تعدادی از پلیمورفیسمهای نواحی ژنی مرتبط با سرطان پروستات

ارتباط تعدادی از بازآزاییهای کروموزومی نیز با سرطان پروستات مشخص شده است که رایجترین آن TMPRSS2-ERG است.ERG یک تنظیم کننده رونویسی است و مطالعات invitro نشان دادهاند که افزایش بیان ERG میتواند منجر به ایجاد سلولهای سرطانی مهاجم پروستات شود (80). ERG و TMPRSS2 حدوداً Mb3 بر روی کروموزوم 22q21 از هم فاصله دارند و ادغام آنها معمولاً بین اگزون 1 یا 2 از TMPRSS2 و اگزون 2، 3، 4 یا 5 از ERG رخ میدهد. این ادغام در اثر جابجاییهای کروموزومی و یا حذف قسمتی از DNA روی میدهد (80).

شکل 13. برخی از عوامل مؤثر بر پیشرفت سرطان پروستات
مطالعات اخیر ارتباط بین فراوانی ادغام TMPRSS2-ERG با سرطان پروستات را در نمونههای مختلف نشان داده است. در 78 نمونه سالم هیچ موردی از ادغام این دو مشاهده نشد. در 84 مورد نمونه هایپرپلاژی خوشخیم پروستات، 2 مورد مشاهده شد که معادل 2/4 درصد است. در 45 نمونه از افرادی که دارای
high-grade prostatic intraepithelial neoplasia بودند 9 مورد مشاهده شد که معادل 20 درصد است. نهایتاً این ادغام در 692 نفر از 1374 موردی که درگیر سرطان پروستات بودند مشاهده شد، که معادل 50 درصد می باشد (81).
احتمالاً ادغام TMPRSS2-ERG انکوژن MYC را فعال میکند که در نزدیکی ناحیه 24q8 بوده که پلیمورفیسمهای متعددی در این ناحیه با سرطان پروستات مرتبط میباشند و بنابراین میتوانند در پیشبینی خطر ابتلا به بیماری کمک کننده باشند (81, 82).
با وجود یافتههای فوق همچنان مطالعات بیشتری بر جمعیتهای مختلف نیاز است تا بتوان ارتباط جهشهای سوماتیک و پلیمورفیسمهای مختلف با سرطان پروستات را آشکار ساخت (70).
ژنوميكس و سرطانهاي دستگاه توليد مثل
بررسي وضعيت بيان ژنها براي شناسايي الگوي تغيير بيان در كل ژنوم، درك بهتري از وقايع ملكولي در انواع بيماريها مانند سرطان را فراهم و امكان تمايز بين انواع بيماريهاي پاسخ دهنده و انواع مقاوم به داروهاي خاص و پيشبيني ميزان سميت و عوارض جانبي دارو را ايجاد ميكند (83, 84). درمان سرطان با روشهاي اشعه درماني و شيمي درماني و مقاومت نسبت به درمان به عنوان اصليترين مشكل كلينيكي مطرح است. عدم پاسخ برخي بيماران به چنين درمانهايي احتمالاً توسط مطالعة الگوي بيان ژني هر بيمار به طور جداگانه قابل پيشبيني ميباشد. اين تكنولوژيهاي جديد در بسياري از انواع سرطانها شامل سرطانهاي دستگاه توليد مثل (سينه، تخمدان، دهانه رحم، پروستات، اندومتر) به خدمت گرفته شدهاند. در رويه كنوني درمان سرطان بدون در نظر گرفتن پاسخ دادن يا مقاومت بيمار، اشعه درماني در مورد همه بيماران به طور يكسان انجام ميگيرد (84).
ژنوميكس سرطان سينه
يافتههاي Microarray در تحقيقات سرطان سينه به طور عمده به دو موضوع اشاره ميكنند؛ اولاً، تومورهاي جداگانه كه از يك عضو منشأ ميگيرند ممكن است بر اساس الگوي بيان ژني و مستقل از مرحله و درجه پيشرفت، در گروههاي متفاوتي طبقه بندي شوند. ثانياً، از يافتههاي بيولوژيكي ناشي از اينگونه طبقه بنديها ميتوان جهت تشخيص استفاده كرد. مطالعات نشان داده است كه تكنولوژي Microarray امكان بررسي رفتار تومور در بافت زنده را فراهم و ارزيابي روش تشخيص و مقاومت دارويي را امكان پذير ميسازد. بررسي بيان هزاران ژن نشان ميدهد كه تفاوت زيادي در بين تومورهايي كه از يك عضو منشأ ميگيرند وجود دارد (85, 86).
به نظر ميرسد كه مهمترين چالش كنوني استفاده از DNA Microarrayدر ارزيابي بيان ژن و تغييرات تعداد رونوشتها در ماده توموري باشد. با وجود اينكه تكنولوژي DNA Microarray در آناليز بيـــــان ژنها پيشرفتي در درمان كلينيكي بيماران سرطان سينه ايجاد نكرده است، اما اطلاعاتي درباره مسيرها و مكانيسمهاي ملكولي در فعاليتهاي بيولوژيكي، فراهم ميكند (86).
ژنوميكس سرطان تخمدان
با وجود سالها تحقيق و پيشرفتهاي قابل ملاحظه در درك مستعد بودن براي سرطان تخمدان، اين سرطان به عنوان علت اصلي مرگ در بين بيماریهاي زنان شناخته شده است. پيشرفتهاي اخير در تكنيکهاي پربازده براي مطالعه ژنوم، تحقيق براي شناسايي بيوماركرها جهت شناسايي زود هنگام و تشخيص بيماري را سرعت بخشيده است (87).
آناليز دادههاي Microarray منجر به شناسايي گروه ژني كه تغييرات بياني آشكار داشتند گرديد. براي جستجوي بيوماركرهاي مراحل اوليه بيماري، تكنيك بررسي بيان ژنها در نمونههاي بيوپسي تومور و كشت اوليه كوتاه مدت كارسينوماي سروزي تخمدان، به كارگرفته شد كه منجر به كشف بيوماركرهاي ملكولي نوين براي تشخيص زود هنگام و درمان گرديد (88).
در مطالعه ديگري يك Oligonucleotide Microarray با پروبهاي مكمل براي بيشتر از 14500 ژن انساني به كار گرفته شد تا تفاوت الگوي بيان ژن در تومورهاي تخمدان پاپيلاري سروزي متاستاتيك در مقايسه با كارسينوماي سروزي اوليه تخمدان بررسي شود (89). آناليز بيان ژنها با Microarray همچنين براي تعيين خصوصيات پيشرفت سرطانهاي سروزي به كار گرفته شد (90). در یک مطالعه، 31 نمونه از بیماران مبتلا به مراحل پيشرفته سرطان تخمدان سروزي، که كمتر از 2 سال و بيشتر از 7 سال زنده مانده بودند، با 3 نمونه اپيتليال تخمدان سالم مقایسه شدند. اين مطالعه الگوهاي بيان ژنهاي مرتبط با گسترش سرطان تخمدان و نتايج آن را شناسايي كرد. در آينده آناليز ژنتيكي دقيقتر و كاملتر ممكن است منجر به كشف ماركرهايي براي تشخيص زود هنگام و پيشبيني الگوهاي بيان، براي منظورهاي درماني شود (90).
با توجه به کاهش روزافزون هزینههای توالی یابی و بررسی ژنوم، افزایش کارایی روشهای موجود، پیشرفتهای علم شیمی و ورود نسلهای جدید توالی یابی احتمالاً در آینده بیمارانی که به مراکز درمانی مراجعه میکنند قبل از معاینه توالی ژنی خود را به همراه خواهند داشت. با این وجود این روند انقلابی نیست که در یک شب اتفاق بیفتد، چون پیشبینی فنوتیپ بر اساس یافتههای ژنوتیپی همچنان با محدودیتهای فراوان روبرو است و همچنین زمان زیادی تا ظهور تکنولوژیهای ژنومیکی با کیفیت مناسب و هزینه کم باقی مانده است (36).
- 1. “A Brief Guide to Genomics”. Genome.gov [Internet]. 2010-11-08 [cited Retrieved 2011-12-03].
- Institute NHGR. “FAQ About Genetic and Genomic Science”. Genome.gov. (2010-11-08). p. Retrieved 2011-12-03.
- Yadav SP. “The wholeness in suffix -omics, -omes, and the word om”. Journal of biomolecular techniques : JBT. 2007;18 (5):277.
- 4. Ankeny RA. Sequencing the genome from nematode to human: changing methods, changing science. Endeavour. 2003;27(2):87-92.
- 5. Holley RW, Apgar J, Everett GA, Madison JT, Marquisee M, Merrill SH, et al. Structure of a ribonucleic acid. Science. 1965;147(3664):1462-5.
- Holley RW, Everett GA, Madison JT, Zamir A. Nucleotide sequences in the yeast alanine transfer ribonucleic acid. Journal of Biological Chemistry. 1965;240(5):2122-8.
- Nirenberg M, Leder P, Bernfield M, Brimacombe R, Trupin J, Rottman F, et al. RNA codewords and protein synthesis, VII. On the general nature of the RNA code. Proceedings of the National Academy of Sciences of the United States of America. 1965;53(5):1161.
- Jou WM, Haegeman G, Ysebaert M, Fiers W. Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein. Nature. 1972;237:82-8.
- 9. Fiers W, Contreras R, Haegemann G, Rogiers R, Van De Voorde A, Van Heuverswyn H, et al. Complete nucleotide sequence of SV40 DNA. Nature. 1978;273(5658):113-20.
- Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, et al. Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. 1976.
- 11. Sanger F. Determination of nucleotide sequences in DNA. Bioscience reports. 1981;1(1):3-18.
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences. 1977;74(12):5463-7.
- Maxam AM, Gilbert W. A new method for sequencing DNA. Proceedings of the National Academy of Sciences. 1977;74(2):560-4.
- “The Nobel Prize in Chemistry 1980”. Nobelprize.org. Nobel Media AB 2013. Web. 11 Apr 2014 [Internet].
- Anderson S, Bankier AT, Barrell BG, De Bruijn M, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. 1981.
- Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462(7276):1056-60.
- McElheny VK. Drawing the map of life: inside the human genome project: Basic Books; 2012. 52 p.
- Barnes B, Dupré J. Genomes and what to make of them: University of Chicago Press: 37; 2009. 37 p.
- Fields S, Song O-k. A novel genetic system to detect protein protein interactions. 1989.
- Chandonia J-M, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311(5759):347-51.
- Kuhn P, Wilson K, Patch MG, Stevens RC. The genesis of high-throughput structure-based drug discovery using protein crystallography. Current opinion in chemical biology. 2002;6(5):704-10.
- Russel P. iGenetics. Eds: Benjamin Cummings, CA. 2002:190-2.
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. Journal of bacteriology. 1998;180(18):4765-74.
- Crotwell PL, Hoyme HE. Advances in whole-genome genetic testing: from chromosomes to microarrays. Current problems in pediatric and adolescent health care. 2012;42(3):47-73.
- Gustashaw KM. Chromosome stains. The ACT cytogenetics laboratory manual. 1991;222.
- Tjio JH, Levan A. The chromosome number of man. Hereditas. 1956;42(1‐2):1-6.
- Speicher MR, Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nature Reviews Genetics. 2005;6(10):782-92.
- oconnor C. Fluorescence in situ hybridization(FISH). Nature education. 2008;1(1):171.
- Chang T-W. Binding of cells to matrixes of distinct antibodies coated on solid surface. Journal of immunological methods. 1983;65(1):217-23.
- Khan A. Genomics and microarray for detection and diagnostics. Acta microbiologica et immunologica hungarica. 2004;51(4):463-7.
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. science. 1999;286(5439):531-7.
- Drake FH, Dodds RA, James IE, Connor JR, Debouck C, Richardson S, et al. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. Journal of Biological Chemistry. 1996;271(21):12511-6.
- Sonoda G, Palazzo J, du Manoir S, Godwin AK, Feder M, Yakushiji M, et al. Comparative genomic hybridization detects frequent overrepresentation of chromosomal material from 3q26, 8q24, and 20q13 in human ovarian carcinomas. Genes, chromosomes and cancer. 1997;20(4):320-8.
- McCutchan TF, Singer MF. DNA sequences similar to those around the simian virus 40 origin of replication are present in the monkey genome. Proceedings of the National Academy of Sciences. 1981;78(1):95-9.
- theisen A. microarray based comparative genomic hybridization. nature education. 2008;1(1):45.
- Katsanis SH, Katsanis N. Molecular genetic testing and the future of clinical genomics. Nature Reviews Genetics. 2013;14(6):415-26.
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic acids research. 2002;30(12):e57-e.
- Eldering E, Spek CA, Aberson HL, Grummels A, Derks IA, de Vos AF, et al. Expression profiling via novel multiplex assay allows rapid assessment of gene regulation in defined signalling pathways. Nucleic acids research. 2003;31(23):e153-e.
- Caspersson T, Zech L, Johansson C. Analysis of human metaphase chromosome set by aid of DNA-binding fluorescent agents. Experimental cell research. 1970;62(2):490-2.
- Pevsner J. Bioinformatics and functional genomics: John Wiley & Sons: 71; 2009.
- Staden R. A strategy of DNA sequencing employing computer programs. Nucleic acids research. 1979;6(7):2601-10.
- Anderson S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Research. 1981;9(13):3015-27.
- Hall N. Advanced sequencing technologies and their wider impact in microbiology. Journal of Experimental Biology. 2007;210(9):1518-25.
- Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols. 2010;2010(6):pdb. prot5448.
- Pop M. Genome assembly reborn: recent computational challenges. Briefings in bioinformatics. 2009;10(4):354-66.
- Chain P, Grafham D, Fulton R, Fitzgerald M, Hostetler J, Muzny D, et al. Genome project standards in a new era of sequencing. Science (New York, NY). 2009;326(5950).
- Stein L. Genome annotation: from sequence to biology. Nature reviews genetics. 2001;2(7):493-503.
- Brent MR. Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nature Reviews Genetics. 2008;9(1):62-73.
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. science. 2001;291(5507):1304-51.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921.
- Michener CM, Ardekani AM, Petricoin III EF, Liotta LA, Kohn EC. Genomics and proteomics: application of novel technology to early detection and prevention of cancer. Cancer detection and prevention. 2002;26(4):249-55.
- Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, et al. Laser capture microdissection. Science. 1996;274(5289):998-1001.
- Jares P, Campo E. Genomic platforms for cancer research: potential diagnostic and prognostic applications in clinical oncology. Clinical and Translational Oncology. 2006;8(3):161-72.
- Chung CH, Levy S, Chaurand P, Carbone DP. Genomics and proteomics: emerging technologies in clinical cancer research. Critical reviews in oncology/hematology. 2007;61(1):1-25.
- Petricoin III EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, et al. Use of proteomic patterns in serum to identify ovarian cancer. The lancet. 2002;359(9306):572-7.
- Mittal V, Nolan DJ. Genomics and proteomics approaches in understanding tumor angiogenesis. 2007.
- Franzén B, Hirano T, Okuzawa K, Uryu K, Alaiya AA, Linder S, et al. Sample preparation of human tumors prior to two‐dimensional electrophoresis of proteins. Electrophoresis. 1995;16(1):1087-9.
- Zvibel I, Brill S, Papa M, Halpern Z. Hepatocyte-derived soluble factors regulate proliferation and autocrine growth factor expression in colon cancer cell lines of varying liver-colonizing capability. Tumor biology. 2000;21(4):187-96.
- Bemis LT, Schedin P. Reproductive state of rat mammary gland stroma modulates human breast cancer cell migration and invasion. Cancer research. 2000;60(13):3414-8.
- Berger SJ, DeVries GW, Carter JG, Schulz DW, Passonneau PN, Lowry O, et al. The distribution of the components of the cyclic GMP cycle in retina. Journal of Biological Chemistry. 1980;255(7):3128-33.
- Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411(6835):375-9.
- Kloth JN, Oosting J, van Wezel T, Szuhai K, Knijnenburg J, Gorter A, et al. Combined array-comparative genomic hybridization and single-nucleotide polymorphism-loss of heterozygosity analysis reveals complex genetic alterations in cervical cancer. BMC genomics. 2007;8(1):53.
- Fodor S, Rava RP, Huang XC, Pease AC, Holmes CP, Adams CL. Multiplexed biochemical assays with biological chips. Nature. 1993;364:555-6.
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nature genetics. 1996;14(4):457-60.
- Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, et al. Gene expression profiles in normal and cancer cells. Science. 1997;276(5316):1268-72.
- Kononen J, Bubendorf L, Kallionimeni A, Bärlund M, Schraml P, Leighton S, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nature medicine. 1998;4(7):844-7.
- Schaid DJ. The complex genetic epidemiology of prostate cancer. Human molecular genetics. 2004;13(suppl 1):R103-R21.
- Siegel R, Ward E, Brawley O, Jemal A. The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA-A CANCER JOURNAL FOR CLINICIANS. 2011;61(4):212-36.
- Baade PD, Youlden DR, Krnjacki LJ. International epidemiology of prostate cancer: geographical distribution and secular trends. Molecular nutrition & food research. 2009;53(2):171-84.
- Witte JS. Prostate cancer genomics: towards a new understanding. Nature Reviews Genetics. 2009;10(2):77-82.
- Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nature genetics. 2007;39(5):631-7.
- Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nature genetics. 2007;39(5):645-9.
- Cheng I, Plummer SJ, Jorgenson E, Liu X, Rybicki BA, Casey G, et al. 8q24 and prostate cancer: association with advanced disease and meta-analysis. European Journal of Human Genetics. 2008;16(4):496-505.
- Haiman CA, Le Marchand L, Yamamato J, Stram DO, Sheng X, Kolonel LN, et al. A common genetic risk factor for colorectal and prostate cancer. Nature genetics. 2007;39(8):954-6.
- Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nature genetics. 2008;40(11):1307-12.
- Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nature genetics. 2008;40(3):310-5.
- Eeles RA, Kote-Jarai Z, Giles GG, Al Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nature genetics. 2008;40(3):316-21.
- Beke L, Nuytten M, Van Eynde A, Beullens M, Bollen M. The gene encoding the prostatic tumor suppressor PSP94 is a target for repression by the Polycomb group protein EZH2. Oncogene. 2007;26(31):4590-5.
- Buckland PR, Hoogendoorn B, Coleman SL, Guy CA, Smith SK, O’Donovan MC. Strong bias in the location of functional promoter polymorphisms. Human mutation. 2005;26(3):214-23.
- Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nature Reviews Cancer. 2008;8(7):497-511.
- Sun C, Dobi A, Mohamed A, Li H, Thangapazham R, Furusato B, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene. 2008;27(40):5348-53.
- Laxman B, Morris DS, Yu J, Siddiqui J, Cao J, Mehra R, et al. A first-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer research. 2008;68(3):645-9.
- Clarke PA, te Poele R, Wooster R, Workman P. Gene expression microarray analysis in cancer biology, pharmacology, and drug development: progress and potential. Biochemical pharmacology. 2001;62(10):1311-36.
- Chin K-V, Kong A-NT. Application of DNA microarrays in pharmacogenomics and toxicogenomics. Pharmaceutical research. 2002;19(12):1773-8.
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747-52.
- Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences. 2001;98(19):10869-74.
- Grisaru D, Hauspy J, Prasad M, Albert M, Murphy KJ, Covens A, et al. Microarray expression identification of differentially expressed genes in serous epithelial ovarian cancer compared with bulk normal ovarian tissue and ovarian surface scrapings. Oncology reports. 2007;18(6):1347.
- Bignotti E, Tassi RA, Calza S, Ravaggi A, Romani C, Rossi E, et al. Differential gene expression profiles between tumor biopsies and short-term primary cultures of ovarian serous carcinomas: identification of novel molecular biomarkers for early diagnosis and therapy. Gynecologic oncology. 2006;103(2):405-16.
- Bignotti E, Tassi RA, Calza S, Ravaggi A, Bandiera E, Rossi E, et al. Gene expression profile of ovarian serous papillary carcinomas: identification of metastasis-associated genes. American journal of obstetrics and gynecology. 2007;196(3):245. e1-. e11.
- Lancaster JM, Dressman HK, Whitaker RS, Havrilesky L, Gray J, Marks JR, et al. Gene expression patterns that characterize advanced stage serous ovarian cancers. Journal of the Society for Gynecologic Investigation. 2004;11(1):51-9.
ژنومیکس و کاربرد آن در تشخیص بیماریها (2)
https://www.chop.edu/centers-programs/division-genomic-diagnostics
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام