بیمار بدحال با هیپرگاماگلوبولینمی شدید
زهرا مولایی دانشجوی کارشناسی ارشد هماتولوژی
یک آقای ۳۰ ساله با سابقه ۶ ماه لنفادنوپاتی پیشرونده و با علائم خستگی، تعریق شبانه، کاهش وزن و تنگی نفس به بخش هماتولوژی اونکولوژی ارجاع داده شد. گزارش زمان ارجاع نشان میداد که حال بیمار به حدی به وخامت گراییده بود که قادر به کار کردن نبود و نمیتوانست فعالیتهای فیزیکی خود را به شدت قبلی انجام دهد.
در معاینه، دیسترس حاد مشهود نبود و بیمار علائم حیاتی نرمال داشت. در معاینه بالینی، آدنوپاتی دوطرفه گردن، زیربغل و کشاله ران دیده شد. شکم بیمار نرم و بدون درد در هنگام لمس بود و هپاتواسپلنومگالی مشهود نبود. مشخص شد که بیمار بهطور منظم سیگار میکشد و هر از گاهی الکل و ماریجوانا مصرف میکند. بیمار سابقه تزریق مواد مخدر و سفر خارجی اخیر نداشت. بر اساس سابقه فامیلی بیمار، مادربزرگ مادری وی مبتلا به لنفوم غیرهوچکین و دو عموزاده او نیز مبتلا به سرطان کبد و کولون تشخیص داده شده بودند.
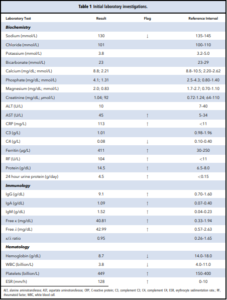
نتایج بررسیهای آزمایشگاهی اولیه وی در جدول ۱ خلاصه شده است. آزمایشها، وجود آنمی، ترومبوسیتوز و هایپرگاماگلوبولینمی چشمگیر را آشکار ساختند. الکتروفورز پروتئینهای سرم و ادرار، گاماپاتی مونوکلونال را مشخص نساخت، پروتئینوری در محدوده نفروتیک اما کراتینین سرم نرمال بود. بررسیهای بیشتر بر روی خون بیمار، التهاب فعال به همراه افزایش سدیمان، پروتئین واکنشگر C و غلظت فریتین و نیز کاهش C4 کمپلمان را نشان داد. دلایل عفونی برای لنفادنوپاتی و التهاب سیستمیک کنار گذاشته شد، زیرا نتایج تست بیمار برای هپاتیت B، هپاتیت C، ویروس اپشتین بار، ویروس نقص ایمنی انسانی و هرپس ویروس انسانی ۸ (HH8) منفی بود. HH8 با رنگآمیزی بافت غدد لنفاوی، سنجش ایمونوفلورسانس و PCR خون محیطی تأیید شد. علیرغم اینکه بیمار وجود هرگونه علائم مفصلی فعال را رد کرد، با توجه به افزایش فاکتور روماتوئید، وجود بیماریهای اتوایمیون زمینهای در نظر گرفته شد. به دلیل نگرانی از وخامت بالینی سریع- به ویژه در ارتباط با عملکرد کلیوی- درمان با پردنیزون (1 mg/kg) آغاز شد و بررسیهای بیشتر برای تعیین علت لنفادنوپاتی ادامه یافت.
بیوپسی از گره لنفی کشاله ران پای چپ، پلاسماسیتوز داخل فولیکولی به همراه هایپرپلازی فولیکولار خفیف بدون افزایش در لنفوسیتها را نشان داد که پاتولوژی غیرلنفومی را مطرح میساخت. منحصر شدن ساخت ایمونوگلوبین به زنجیره سبک مشاهده نشد (و بعداً در فلوسایتومتری نیز تأیید شد)؛ با این حال افزایش پلاسماسلهای IgG4 مثبت وجود داشت. تشخیص افتراقی در این مرحله، بین واریانت پلاسماسلی بیماری کاسلمن (CD) و بیماری مرتبط با IgG4 بود، اگرچه برای تأیید هر یک از این دو تشخیص، یافتههای بالینی بیشتری موردنیاز بود. یک بیوپسی کلیه نیز برای تعیین اینکه آیا سندرم نفروتیک به علت یک بیماری اتوایمیون زمینهای رخ داده است یا اینکه تظاهری از اختلال لنفوپرولیفراتیو است، انجام شد. بیوپسی، گلومرولونفریت بهواسطه کمپلکس ایمنی بدون اسکلروزیس چشمگیر را نشان داد که نشان میدهد علائم کلیوی به اختلال اولیه بافت همبند یا اختلال اتوایمیون مرتبط نیست. بررسیهای بیشتر، از جمله منفی بودن تست بیمار برای آنتیبادیهای ضد هستهای، ضد پپتیدهای سیترولینه حلقوی، ضد سیتوپلاسم نوتروفیل و ضد غشای پایه گلومرول؛ کنار گذاشتن این بیماریها را تأیید کرد.
برای درک یافتههای بیوپسی گره لنفی، اندازهگیری اینترلوکین ۶ (IL-6) و زیرگونههای IgG در خون انجام شد زیرا IL-6 غالباً در بیماری کاسلمن افزایش مییابد و زیرگونههای IgG برای تأیید تشخیص بیماری مرتبط با IgG4 مفید هستند. اگرچه IgG4 تا 1.022 g/dL افزایش یافته بود (بازه مرجع: 0/086-0/004 g/dL)، اما به دلیل اینکه همه زیرگونههای IgG افزایش یافته بودند و نسبت IgG4:IgG در بیوپسی گره لنفی افزایش چشمگیری نداشت، بیماری مرتبط با IgG4 کنار گذاشته شد. مقدار IL-6 پلاسما تا 93.6 pg/mL (بازه مرجع 11.9-0 pg/mL) افزایش یافته بود. با در نظر گرفتن افزایش غلظت IL-6 در کنار ویژگیهای بالینی و پاتولوژیک، تشخیص نهایی، بیماری کاسلمن چندمرکزی بود.
بهمحض قطعی شدن تشخیص، اضافه کردن سرکوبکنندههای ایمنی به رژیم درمانی استروئیدی بیمار، مورد بحث قرار گرفت. در نهایت ریتوکسیمب (آنتیبادی مونوکلونال ضد CD20) بهجای سیلتوکسیمب (آنتیبادی مونوکولنال ضد IL-6) برای بیمار در نظر گرفته شد؛ زیرا سیلتوکسیمب میبایست بهصورت پیوسته و بدون مشخص بودن زمان پایان مصرف دارو، مورد استفاده قرار میگرفت. تقریباً ۲ ماه بعد از درمان با ریتوکسیمب و پردنیزون، بهبود لنفادنوپاتی و عدم بروز علائم جدید گزارش شد. تستهای آزمایشگاهی از قبیل هموگلوبین، پروتئین تام و ترشح پروتئین در ادرار و نیز مارکرهای التهابی، بهطور پیشروندهای به سمت مقادیر نرمال حرکت میکردند.
| سؤالات مهم:
1) تشخیص افتراقی هایپرگاماگلوبینمی پلیکلونال چیست؟ 2) برای تعیین علت هایپرگاماگلوبینمی پلیکلونال، چه بررسیهای دیگری باید انجام شود؟ 3) اهمیت اندازهگیری سایتوکاین در اختلالات لنفوپرولیفراتیو چیست؟ |
شرح کیس
بیماری کاسلمن (CD)، یک اختلال هماتولوژیک نادر است که اولین بار در سال ۱۹۵۶ توسط کاسلمن و همکاران بهعنوان یک توده بزرگ شبه تیموما که در گره لنفی مدیاستینوم قدامی قرار گرفته بود، توصیف شد. مشخص شده است بیماری کاسلمن که تحت عنوان بیماری لنفوپرولیفراتیو پلیکلونال نیز شناخته میشود، گروه هتروژنی از بیماریهای لنفوپرولیفراتیو با اتیولوژی غالباً نامعلوم است. تحقیقات اخیر نشان میدهند که برخی زیرگروههای بیماری کاسلمن با جابجاییهای کلونال ژنتیکی مرتبط هستند، اما این یافتهها باید در مطالعات اعتبارسنجی بزرگتر، اثبات شوند. به لحاظ تاریخی، بیماری کاسلمن با توجه به تعداد گرههای لنفی درگیرشده، به بیماری کاسلمن تکمرکزی (unicentric CD; uCD) و بیماری کاسلمن چندمرکزی (multicentric CD; mCD) طبقهبندی میشود. uCD به یک گره لنفی محدود میشود (رایجترین موقعیت در مدیاستینوم، گردن یا شکم است) و معمولاً با جراحی قابل درمان است اما mCD با درگیری چند گره لنفی مشخص میشود که باعث التهاب سیستمیک، آناسارکا و متأثر شدن چند ارگان میگردد، بنابراین mCD با وضعیت بسیار تهاجمیتر و پروگنوز ضعیفتری همراه است
هایپرگاماگلوبینمی یک تظاهر شایع بیماری کاسلمن بهعنوان یک بیماری لنفوپرولیفراتیو است. هایپرگاماگلوبینمی عموماً با ایمونوگلوبینهای اضافی تولیدشده توسط یک کلون پلاسماسل به دلیل بدخیمی، همراهی دارد. مثالهایی از این بدخیمیهای گاماپاتی مونوکلونال عبارتند از: مالتیپل میلوما، آمیلوئیدوز اولیه و ماکروگلوبولینمی والدنشتروم. در مقابل، گاماپاتیهای همراه با بیماری کاسلمن عموماً ماهیت پلیکلونال دارند زیرا لنفوسیتوز زمینهای به یک جمعیت کلونال محدود نشده است. گاماپاتیهای پلیکلونال غالباً از طریق گسترش یافتن باند گاما در الکتروفورز پروتئینهای سرم مشخص میشوند. هایپرگاماگلوبولینمی پلیکلونال همانند دیگر تظاهرات بیماری کاسلمن، غیراختصاصی بوده و میتواند مرتبط به طیفی از بیماریهای مختلف مانند بیماری کبدی، بیماری بافت همبند، اختلالات هماتولوژیک، عفونتها و بدخیمیهای غیرهماتولوژیک باشد. از آنجایی که بیماری کاسلمن یک بیماری نادر است، انجام بررسیهای گسترده بر روی موارد مشکوک به این بیماری دارای اهمیت است تا بتوان دیگر بیماریهای مشابه را کنار گذاشت.
به لحاظ بافتشناسی، بیماری کاسلمن به انواع هیالین واسکولار، پلاسماسلی یا مختلط طبقهبندی میشود. در نوع هیالین واسکولار، مراکز زایا در فولیکولها تحلیل رفته یا وجود ندارند و ناحیه جبهای دارای الگوهای متحدالمرکز «پوست پیازی» است. در نوع پلاسماسلی، گرههای لنفی ساختار خود را حفظ کردهاند و دارای فولیکولهای هایپرپلاستیک و پلاسماسیتوز پلیکلونال در نواحی داخل فولیکولی هستند. نوع مختلط، ترکیبی از ویژگیهای هیالین واسکولار و پلاسماسلی را نشان میدهد. بخش عمده uCDها از نوع هیالین واسکولار میباشند و نوع پلاسماسلی، هیستولوژی غالب در mCD است.


به لحاظ بالینی، mCD با علائم «کلاسیک» لنفادنوپاتی، مانند تب، کاهش وزن و خستگی بروز مییابد و یافتههای آزمایشگاهی رایج آن عبارتند از آنمی، هایپرگاماگلوبولینمی و هایپوآلبومینمی. باید توجه داشت که تعیین وضعیت HHV8 در بیماران mCD حیاتی است زیرا mCD وابسته به HHV8 یک علت ویروسی اثباتشده است و بر این اساس درمان میشود. در مقابل، mCD با HHV8 منفی، ماهیت آیدیوپاتیک دارد و با کنار گذاشتن بیماریهایی با تظاهرات بالینی و یافتههای آزمایشگاهی مشابه، تشخیص داده میشود. با وجود اینکه بررسی هیستوپاتولوژیک بیوپسی گره لنفی در تشخیص mCD آیدیوپاتیک (iMCD)، نقش محوری دارد، اما بررسی تستهای آزمایشگاهی نیز برای کنار گذاشتن سایر اختلالات با ویژگیهای مشابه، ضروری است. در واقع، اهمیت بررسیهای هیستوپاتولوژیک و بیوشیمیایی، در معیارهای تشخیصی مورد توافق iMCD انعکاس یافته است. بر اساس مرور ۲۴۴ کیس بالینی و ۸۸ نمونه بافتی توسط گروهی از پزشکان و پاتولوژیستها، اولین معیار تشخیصی برای iMCD، وجود لنفادنوپاتی چندمرکزی با شواهد هیستوپاتولوژیک، دستکم ۲ تغییر بالینی یا آزمایشگاهی و کنار گذاشتن اختلالات مشابه iMCD تعیین شد. با این وجود، درمان iMCD همچنان بهطور آشکاری متغیر است که دلیل آن اندک بودن تحقیقات انجام شده بر روی کارآمد بودن درمانها و فقدان رویکرد استاندارد برای نظارت بر پاسخ بیمار به درمان است.
حتی در صورت عدم وجود ضوابط تشخیصی الزامی، غلظتهای افزایشیافته سایتوکاینها– بهطور خاص اینترلوکین ۶ (IL-6)- نقش اساسی در پاتوژنز اکثر (اما نه همه) موارد بیماری کاسلمــن دارد. در حالت نرمال، IL-6 از لنفوسیتهای T و ماکروفاژها ترشح میشود تا پاسخ ایمنی را در برابر محرکهای ایمنولوژیک تحریک نماید، به علاوه IL-6 فرایندهای متعددی را تحریک و تقویت میکند، مانند رشد لنفوسیتهای B، آنژیوژنز، مهار لنفوسیتهای T تنظیمی، تحریک لنفوسیتهای T کمکی و التهاب. در بیماری کاسلمن، ترشح تنظیمنشده و خارج از کنترل IL-6 از لنفوسیتهای B باعث لنفادنوپاتی مشخص و علائم مربوطه میشود. اهمیت IL-6 در پاتوژنز بیماری کاسلمن با مشاهده اینکه کاهش غلظت IL-6 باعث بهبود علائم میشود و اینکه مسدود ساختن سیگنالینگ IL-6 با استفاده از دارو میتواند علائم را تسکین دهد، اثبات شد. از این رو، تحقیقات بر بررسی درمان ضد IL-6 برای درمان بیماری کاسلمن متمرکز شده است.
اخیراً FDA به سیلتوکسیمب که یک آنتیبادی مونوکلونال کایمریک موشی- انسانی است، برای درمان بیماران مبتلا به iMCD با HHV8 و HIV منفی، تأییدیه داده است که اولین درمان تأییدشده در ایالات متحده امریکا برای iMCD است. تأییدیه FDA بر اساس یک کارآزمایی تصادفیشده دوسو کور صادر شده است که اثبات کرد بیش از ۳۰ درصد بیماران iMCD، پاسخ کامل یا نسبی به سیلتوکسیمب نشان دادهاند، در حالی که در گروه دارونما، میزان آن صفر درصد بوده است. افزون بر این یافتهها، Casper و همکاران ثابت کردند که پروتئین واکنشگر C میتواند یک مارکر مناسب برای پایش بیمارانی که در حال درمان با سیلتوکسیمب هستند، باشد و Fajgenbaum و همکاران، ارزش هدف قرار دادن افکتورهای PI3K/Akt/mTOR را در بیماران مبتلا به iMCD مقاوم به مهار IL-6 نشان دادند.
این نتایج نویدبخش، به همراه افزایش اطلاعات ما از این بیماری نادر، پایههایی را برای گروههایی مانند شبکه تعاملی بیماری کاسلمن (Castleman Disease Collaborative Network; https://cdcn.org) فراهم میآورد تا بر روی استانداردسازی درمان و بهبود پیامدهای بیماران کار کنند.
| نکاتی که باید به خاطر سپرد:
– بیماری کاسلمن یک اختلال لنفوپرولیفراتیو نادر است که تشخیص قطعی آن نیازمند ترکیبی از بررسیهای بالینی، هیستوپاتولوژیکال و آزمایشگاهی است. – بیماری کاسلمن میتواند تکمرکزی یا چندمرکزی باشد و نوع چندمرکزی با وضعیت بالینی تهاجمیتر و پروگنوز ضعیف همراه است و با علائم سیستمیک شدیدتری بروز مییابد. – تعیین وضعیت HHV8 برای مدیریت مناسب بیماران مبتلا به بیماری کاسلمن چندمرکزی ضروری است. – هایپرسایتوکاینمی، پاتوژنز محوری بیماری کاسلمن است و در بیشتر تحقیقات، IL-6 بهعنوان سایتوکاین اصلی دخیل در پاتوژنز شناخته شده است، از این رو غلظت IL-6 موجود در گردش خون میتواند تشخیص را اثبات کند. – مهار IL-6 برای استفاده بالینی تأیید شده است و برای برخی بیماران مبتلا به کاسلمن مفید بوده است. |
برگردان از:
A Case of Rapid Deterioration with Marked Hypergammaglobulinemia
Clinical Chemistry 66:11 (2020)
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام