
فنیلکتونوریا
دکتر داریوش فرهود1،2؛ سمیه عمادی کوچک3؛
1: دانشکده بهداشت، دانشگاه علوم پزشکی تهران، تهران، ایران.
2: گروه علوم پایه/ اخلاق، فرهنگستان علوم پزشکی ایران، تهران، ایران.
3: کلینیک ژنتیک دکتر فرهود، تهران، ایران.
پیشگفتار
بیماریهای متابولیکی یا بیوشیمیایی تکژنی، معمولاً با عنوان «خطاهای ذاتی متابولیسم» شناخته میشوند. بزرگترین گروه از این نوع بیماریها، اختلالات مربوط به متابولیسم اسیدآمینه و پپتیدها هستند، که دارای زیرمجموعههای بسیاری میباشند. فنیلکتونوریا (Phenylketonuria) یک اختلال متابولیسم فنیلآلانین یا تیروزین است که به اختصار PKU گفته میشود و در گروه خطاهای ذاتی متابولیسم دستهبندی میگردد (3).
فنیلکتونوری، شایعترین اختلال متابولیسم اسیدآمینه (5) و اولین اختلال ژنتیکی در انسان است که عامل ایجاد آن (در سال 1953 توسط Jervis)، یک نقص آنزیمی خاص شناخته شده است (3). در این اختلال نقص آنزیم فنیلآلانین هیدروکسیلاز (PAH)، یعنی آنزیم موردنیاز برای تبدیل فنیلآلانین به تیروزین، وجود دارد که منجر به ایجاد یک «سد ژنتیکی» در مسیر متابولیسم میگردد (3). در صورت عدم تشخیص و درمان این اختلال، بیماران PKU به دلیل مسمومیت عصبی، در اثر افزایش فنیلآلانین در بدن، مبتلا به رشد شناختی ناقص پس از تولد (6)، آسیبهای ذهنی شدید و حملات مغزی میشوند (3).
اسامی شناختهشدة دیگر بیماری PKU عبارتند از: نقص فنیلآلانین هیدروکسیلاز (نقص PAH)، اولیگوفرنی فنیلپیروویکا (Oligophrenia Phenylpyruvica) و بیماری فولینگ (Fölling disease)(6). دلیل نامگذاری اخیر، شناسایی نوع خاصی از عقبافتادگی ذهنی شدید توسط دکتر آزبورن فولینگ در سال 1934 در نروژ میباشد (7). فولینگ نشان داد که اسید فنیلپیروویک در ادرار کودکان با عقبافتادگی ذهنی، آزاد شده و بر اساس مشاهدات خود، بیماری «الیگوفرنی فنیلپیروویک» را بهعنوان یک اختلال ژنتیکی متابولیسم جدید توصیف کرد (7, 8).
انواع هایپر فنیلآلانینمی (HPA)
از آنجا که نقص در آنزیم فنیلآلانین هیدروکسیلاز (PAH) منجر به افزایش اسید آمینه Phe در بدن بیمار میگردد، هر میزان از افزایش فنیلآلانین (HPA) میتواند فنوتایپ «فنیل کتونوریک» محسوب شود. بر این اساس، طبقهبندی پیشنهادی اولیه توسط Kayaalp و همکاران وی در سال 1997 بدین شرح بوده است: دسته یک، PKU (فنیلکتونوریا) که میزان غلظت فنیلآلانین پلاسما در آنها بیش از 1000 µmol/L میباشد. دسته دو، Non-PKU HPA که میزان Phe پلاسما در آنها عموماً بالاتر از 120 و پایینتر از 1000 میکرومولار است. دسته سه، PKU متغیر، شامل افرادی که در هیچکدام از دو دسته پیشین قرار نمیگیرند (9, 10). این طبقهبندی در جدول شماره 1 آورده شده است.
جدول 1. دستهبندی انواع بیماری PKU برحسب میزان PAH
| دستهبندی | میزان فنیلآلانین در پلاسما |
| PKU (فنیلکتونوریا) | >1000 µmol/L |
| Non-PKU HPA (هایپرفنیلآلانینمی غیر فنیلکتونوری) | 1000 – 120 µmol/L |
| PKU متغیر | متغیر |
در طبقهبندی پیشنهادی دیگر توسط Guldberg و همکاران وی در سال 1998، این اختلال بر اساس میزان تحمل Phe در یک روز، پیش از 5 سالگی به چهار دستة PKU کلاسیک (با عدم فعالیت کامل یا نزدیک به کامل PAH)،PKU متوسط، PKU خفیف و هایپرفنیلآلانینمی خفیف (یا MHP) تقسیم میشود (جدول 2) (9, 11).
جدول 2. دستهبندی انواع بیماریPKU بر اساس میزان تحمل فنیلآلانین در یک روز
| دستهبندی | میزان تحمل فنیلآلانین در یک روز |
| PKU کلاسیک | <250-350 mg |
| PKU متوسط | 350-400 mg |
| PKU خفیف | 400-600 mg |
| هایپرفنیلآلانینمی خفیف | <600 µmol/L (10 mg/dL) |
نشانهها و تظاهرات بالینی
فنیلکتونوری، یک اختلال ژنتیکی است که در اثر جهشهای متفاوت در ژن PAH ایجاد میشود. سد آنزیمی ایجادشده در بدن، موجب کمبود تیروزین و در نتیجه کاهش تولید ملانین میگردد، بنابراین کودکان مبتلا اغلب موهای روشن و چشمان آبی دارند (3)، با این حال شواهد کافی موجود است که با بالا رفتن سن کودک رنگ موها تیرهتر میگردد (12)، بهعلاوه بخشهایی از مغز که معمولاً دارای رنگدانه هستند، مانند بخش تیرة مغز (Substantia nigra)، نیز رنگ خود را از دست میدهند (3).
فنوتیپهای مشاهدهشده در بیماران مبتلا به PKU درماننشده، در مقالات متفاوت، بسیار متنوع گزارش شده است؛ اگزما و حملات مغزی در افرادی که عقبافتادگی شدید دارند بیشتر از بیماران دیگر با عقبماندگی خفیف دیده میشوند (12). آزاد شدن اسید فنیلپیروویک و دیگر متابولیتها در ادرار (3) موجب ایجاد بوی غیرعادی کپکزدگی بدن و ادرار و همچنین بیماریهای پوستی (از جمله اگزما) میشود (9). نوار مغزی (انسفالوگراف) در 79% بیماران غیرطبیعی میباشد (12) و عوارض عصبشناختی و رفتاری از جمله افسردگی، اضطراب، فوبیا، مشکلات تمرکزی و انزوای اجتماعی (13)، و مشکلاتی از قبیل لرزش، فلج پا و فلج نیمه بدن در دهههای بعدی زندگی بیماران درماننشده، مشاهده گردیده است (9).

شکل 1. پسر مبتلا به فنیلکتونوری، همراه با موهای بور و کمبود رنگدانه در چشمها (3)
فنیلکتونوری درماننشده، میتواند از جمله اختلالات متابولیکی مادری باشد که موجب انتقال خطر به جنین میگردد. سطح بالای فنیلآلانین در سرم خون مادر باردار، به آسیبهای جدی جنین، از جمله عقبافتادگی ذهنی، ناهنجاریهای ساختاری همچون میکروسفالی و نقایص مادرزادی قلبی منجر خواهد شد (3). نوزادان دارای نقص PAH، معمولاً بدون علائم ظاهری و فیزیکی میباشند، اما در مواردی وزن پایین و کوچک بودن اندازه دور سر نوزاد در هنگام تولد دیده شده است (9).
شیوع بیماری
بر اساس غربالگری انجام شده با استفاده از لکه خون نوزادان، در سالهای 2005 تا 2011 در انگلستان، میزان شیوع PKU یک نفر در هر 10 هزار تولد میباشد (3). شیوع نقص HPA در برخی از جمعیتهای متفاوت، که بین سالهای 1982 تا 2015 بررسی شدهاند، در جدول زیر آمده است.
جدول 3. شیوع نقص HPA در جمعیتهای متفاوت ایران طی سالهای 1980 تا 2015 میلادی (به ترتیب افزایش فراوانی)
| جمعیت | مقدار نمونه | میزان نقص PAH
در 10 هزار تولد زنده |
سال |
| ایران (استان مازندران) (14) | 407244 | 0/66 | 2007-2015 |
| ایران (استان تهران) (15) | 8633 | 1/1 | 1982 |
| ایران (استان فارس) (16) | 175234 | 1/6 | 2004-2007 |
| ایران (استان لرستان) (17) | 384993 | 1/91 | 2006-2016 |
جدول 4. شیوع نقص HPA در برخی جمعیتهای دنیا طی سالهای 1980 تا 2016 میلادی
(به ترتیب افزایش فراوانی)
| جمعیت | مقدار نمونه | میزان نقص PAH
در 10 هزار تولد زنده |
سال |
| ژاپن (18) | 148 | 0/07 | 1988 |
| فنلاند (19) | – | 0/1 – 0/05 | 2016 |
| چین (20) | 17961826 | 0/83 | 2000-2007 |
| ایالات متحده امریکا (21) | 40320849 | 0/84 | 1996-2005 |
| آلمان (22) | 1000000~ | 1 | 1969-1984 |
| ایرلند و اسکاتلند (23) | 550 | 2/22 | 1994-1995 |
| ترکیه (24) | 20979 | 3/84 | 1983-1985 |
مطالعات مربوط به شیوع این بیماری در ایران، تا بحال در چند نمونهبرداری صورت گرفته است. در مطالعهای در سال 1982 در تهران، از 8633 نوزاد متولدشده در آن سال، 7 نوزاد مبتلا به هایپرفنیلآلانینمی مینور و یک مورد مبتلا به هایپرفنیلآلانینمی شدید با میزان بیشتر از 20 گرم درصد بود (15). مطالعه انجام شده بین سالهای 2004 تا 2007 در استان فارس، میزان شیوع PKU را در بین 175234 نوزاد، 1/7 در هر 10 هزار دختر و 1/5 در هر 10 هزار پسر گزارش کرده است (16). در این میان شیوع فنیلکتونوری شدید حدوداً 3 نفر در هر 100 نفر میباشد (16). جدیدترین مطالعات انجامشده در مورد میزان شیوع این بیماری، مربوط به استانهای مازندران و لرستان میباشد که به ترتیب در استان مازندران تعداد 407244 نوزاد متولدشده بین سالهای 2007 تا 2015 بررسی شده و شیوع به میزان 0/66 در 10 هزار نفر تخمین زده شده است (14) و در استان لرستان طی سالهای 2006 تا 2016، در مجموع تعداد 384999 نوزاد بررسی شده و در این میان بیشترین میزان شیوع در سال 2014، با نرخ 3/86 در 10 هزار تولد محاسبه شد (17).
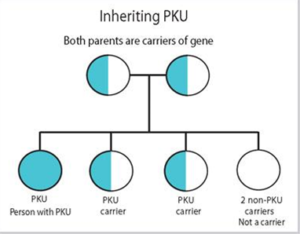
سببشناسی بیماری
انواع متفاوت PKU و نیز هایپرفنیلآلانینمی در اثر جهش در ژن PAH، واقع در کروموزوم 12، ناحیة 12q23.2 (OMIM#612349) ایجاد میگردد. بیش از 950 نوع جهش متفاوت از ژن فنیلآلانین هیدروکسیلاز در مبتلایان به PKU شناخته شده است (25). در این میان انواع جهشهای بدمعنی، بیمعنی، جهشهای واقع در ناحیة برش، حذفها و اضافه شدنهای تغییر چهارچوب، حذفهای داخل چهارچوب و نیز جهشهایی که قابل دستهبندی نبوده و ممکن است در پردازش یا ثبات mRNA مؤثر باشد وجود دارد (11).نقص فنیلآلانین هیدروکسیلاز، یک اختلال ژنتیکی متابولیسم است که الگوی وراثتی اتوزومی نهفته را نشان میدهد.
تشخیص بیماری
بهطور کلی، غربالگری نوزادان، در ابتدا در دهة 1960 میلادی و با بیماری PKU آغاز شد و تاکنون این غربالگری برای طیف وسیعی از بیماریهای متابولیکی و همچنین تست شنوایی گسترش یافته است (3). این بیماری جایگاه ویژهای در تاریخ مطالعة بیماریهای متابولیک کسب کرده است، نهتنها به دلیل آنکه رایجترین اختلال ژنتیکی متابولیسم اسیدآمینه است، بلکه اولین علت خاص شناختهشده برای عقبماندگی ذهنی است و اولین بیماری ژنتیکی جدی است که بهطور مؤثری درمان شد (25).
تشخیص این اختلال، با استفاده از لکهگذاری خون از پاشنة پای نوزاد به روش تست گاتری (Guthrie)، در حدود 6 یا 7 روزگی میباشد(26). اگر نتیجة تست گاتری غیرطبیعی باشد، تست اندازهگیری سطح فنیلآلانین در نمونه خون نوزاد صورت خواهد گرفت (3).
روش غربالگری با لکهگذاری خون نوزاد، مدیون رابرت گاتری، میکروبشناس امریکایی است که نوزاد 15 ماهة خواهر (یا برادر) وی در سال 1958 مبتلا به PKU تشخیص داده شد. وی با استفاده از یک تست مهاری باکتریایی، روشی را توسعه داد که میتوانست سطوح بالای فنیلآلانین را در خون، آن هم در مدت کوتاهی پس از تولد نوزاد تشخیص دهد. گاتری از سال 1961، پیشگام این روش شد (3, 26). او برای این تست از کاغذهای صافی استفاده کرد که لکههای خون میتوانستند بهآسانی روی آنها جمع شده و انتقال یابند- که امروزه هنوز هم این روش مورد استفاده قرار میگیرد.- وی با فشارهای اقتصادی دستوپنجه نرم کرد تا روش خود را با هزینة اندکی معرفی کند (3).
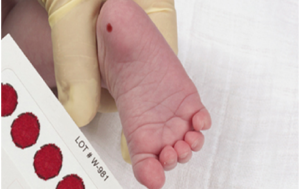
شکل 3. روش غربالگری گاتری با لکهگذاری خون از پاشنه پای نوزاد (2)
درمانهای نشانهای بیماری
همانگونه که در بخشهای پیش گفته شد، جهش در آنزیم PAH موجب اختلال در مسیر متابولیسم تبدیل فنیلآلانین به تیروزین در کبد میگردد. آنزیم PAH در این مسیر نیازمند کوفاکتور BH4 (تتراهیدروبیوپترین) است؛ بنابراین هرگونه نقص در PAH و یا تولید یا بازیابی BH4 میتواند منجر به هایپرفنیلآلانینمی گردد (27).
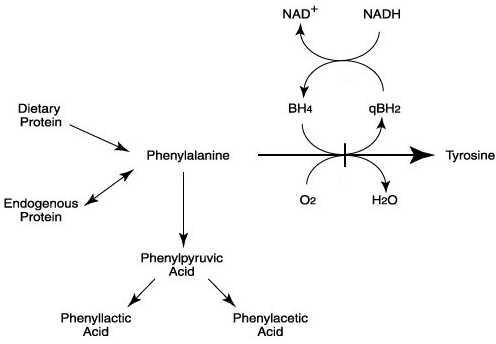
تاکنون تنها روش درمانی PKU، یک رژیم غذایی سخت، کاهشدهنده مصرف فنیلآلانین بوده است (4). برای پیشگیری از بروز عوارض بیماری، آغاز درمانهای تغذیهای باید تا قبل از سومین هفته زندگی نوزاد باشد (27). رژیم غذایی PKU شامل محدود کردن پروتئینهای طبیعی در رژیم غذایی و تکمیل آن با فرمولهای پزشکی خاص است که با تأمین ویتامینها، مواد معدنی و تمام اسیدآمینهها، به غیر از Phe، انجام میشود (4). هدف از رژیمهای تغذیهای، آن است که حداقل به مدت 10 سال اول زندگی، غلظت پلاسمایی فنیلآلانین را در محدودة 120 تا 360 میکرومولار حفظ کند (27). در گذشته، پیشنهاد میشد که این رژیم محدودکننده را میتوان در سنین 4، 6 یا 12 سالگی بدون عوارض جانبی قطع کرد (27)؛ اما از آنجا که سطح بالای فنیلآلانین برای رشد مغز سمی و زیانبار است، زنان مبتلا به PKU در دوران بارداری و حتی پیش از آن، بهتر است از یک رژیم تغذیهای با فنیلآلانین پایین تبعیت کنند (3).
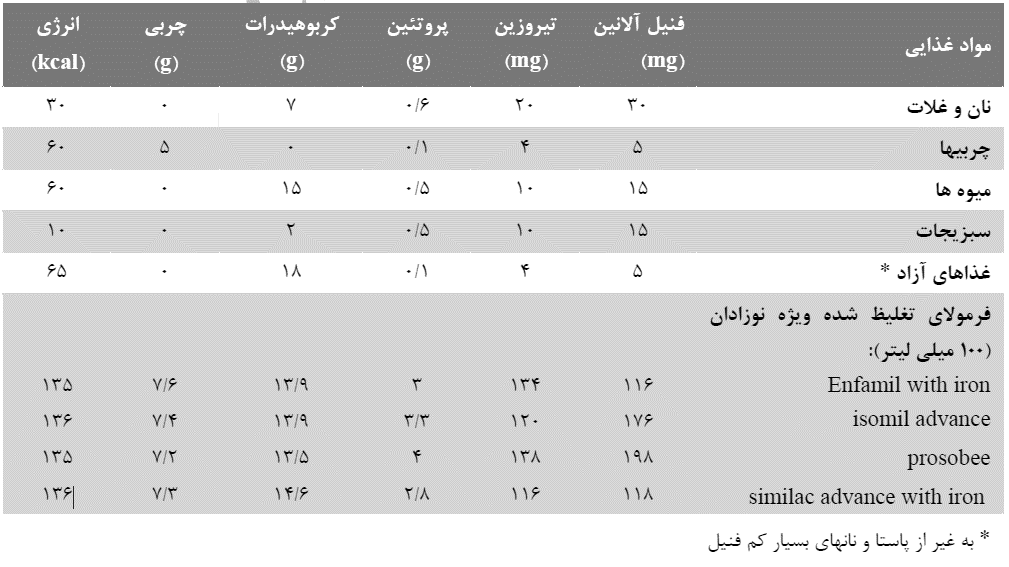
جدول 5. مقادير متوسط مواد مغذي موجود در ليست جانشيني و رژيمهاي غذايي مبتلايان به فنيلكتونوريا (27)
رویکردهای نوین تغذیهای در درمان PKU، که در حال حاضر همراه با رژیم تغذیهای داده میشود، شامل غذاهای خوشطعمتر با استفاده از فراوردههای GMP (گلیکوماکروپپتید)، کاربرد LNAA (اسیدآمینههای خوراکی بزرگ) یا BH4 (کوفاکتور هیدروبیوپترین) میباشند که میتوانند نیاز به محدود کردن فنیلآلانین در رژیم غذایی را کاهش دهند (4).
GMP یک پروتئین طبیعی است که از آب پنیر گرفته میشود و دارای میزان پایین فنیلآلانین و منبعی عالی از پروتئین برای بیماران PKU میباشد. این پروتئین موجب ارتقای طعم، تنوع و غنای غذا میگردد.
مکملهای خوراکی LNAA اسیدآمینههای بزرگی هستند که با انتقال دهندة فنیلآلانین در سیستم معده -رودهای و سد خونی مغز، رقابت کرده و منجر به کاهش جذب و ورود Phe به مغز میگردند. این کاهش غلظت فنیلآلانین مغزی موجب عملکرد روانی- عصبی بهتر میشود (4).
BH4ها و انواع سنتزشدة خوراکی آن از قبیل ساپتوپترین هیدروکلراید، مولکولهای چپرونی هستند که در تاشدگی صحیح و پایداری آنزیم PAH نقش دارند. استفاده از این چپرونهای دارویی، برای پروتئینهای جهشیافته میتواند روشی جدید در درمان بسیاری از بیماریهای ژنتیکی باشد که منجر به بدتاخوردگی پروتئینهای خاصی میشوند. بهطور کلی، درمان با ساپتوپترین، موجب کاهش چشمگیر (حداقل 30%( در غلظت فنیلآلانین خون و افزایش تحمل آن در رژیم غذایی میگردد (4).
این درمانهای تغذیهای جایگزین (شامل BH4 و LNAA) شناخت فیزیوپاتولوژی بیشتری برای علائم روانعصبشناختی (نوروسایکولوژیک) را تأمین میکند. البته از آنجایی که رژیم محدودکنندة فنیلآلانین برای همة بیماران مفید و مؤثر است، GMP بهعنوان منبع پروتئین طبیعی حاوی مقادیر بسیار کم Phe، مفید میباشد (28).
دیگر راهکارهای درمانی:
آنزیمدرمانی؛ که میتواند توسط جایگزینی آنزیمی بهجای PAH و یا جانشینسازی آنزیم فنیلآلانین آمونیا لیاز (PAL) صورت گیرد. پیوند کبد برای بیماران مبتلا به PKU یک گزینة درمانی محسوب نمیشود (4).
سلولدرمانی هدایتشده؛ از دیگر درمانهای تحت مطالعه است که شامل انبوهسازی مجدد کبد با سلولهای بیانکنندة PAH پس از انتقال سلولهای بنیادی کبدی یا سلولهای خونساز میباشد. البته این روش درمانی برای PKU در مدلهای حیوانی با مزیت انتخابی سلولهای گیرنده، گزارش شده است (4).
ژندرمانی؛ برای معالجة PKU کانون توجه گروههای پژوهشی متعددی در دو دهة اخیر بوده است. در مدل موشی PKU، پیشرفتهای مهمی با استفاده از ژن مربوط به آدنوویروس که به داخل کبد هدایت شده، انجام گرفته است؛ بهعلاوه، این مطالعات نشان داده که ژندرمانی میتواند بهطور موفقیتآمیزی به بافتهای غیرکبدی از جمله عضلات فرستاده شود (4).
منابع:
- 1. Park-medlab. [Available from: http://park-medlab.com/index.php/for-doctors.
- 2. PKU and G6PD: zinoteb; 2016 [Available from: http://zinoteb.com/%D9%81%D9%86%DB%8C%D9%84-%DA%A9%D8%AA%D9%88%D9%86%D9%88%D8%B1%DB%8C-%D9%88-g6pd/.
- 3. Turnpenny PD, Ellard S. Emery’s Elements of Medical Genetics E-Book: Elsevier Health Sciences; 2016.
- 4. Strisciuglio P, Concolino D. New strategies for the treatment of phenylketonuria (PKU). Metabolites. 2014;4(4):1007-17.
- 5. Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. The Lancet. 2010;376(9750):1417-27.
- 6. McKusick VA. OMIM # 261600
PHENYLKETONURIA; PKU 6/4/1986 [updated 9/2/2015.
- 7. Fölling A. Excretion of phenylpyruvic acid in urine as a metabolic anomaly in connection with imbecility. Nord Med Tidskr. 1934;8:1054-9.
- 8. Følling I. The discovery of phenylketonuria. Acta Paediatrica. 1994;83(s407):4-10.
- 9. Mitchell JJ, Trakadis YJ, Scriver CR. Phenylalanine hydroxylase deficiency. Genetics in Medicine. 2011;13(8):697-707.
- 10. Kayaalp E, Treacy E, Waters PJ, Byck S, Nowacki P, Scriver CR. Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotype-phenotype correlations. The American Journal of Human Genetics. 1997;61(6):1309-17.
- 11. Guldberg P, Rey F, Zschocke J, Romano V, François B, Michiels L, et al. A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. The American Journal of Human Genetics. 1998;63(1):71-9.
- 12. Paine RS. The variability in manifestations of untreated patients with phenylketonuria (phenylpyruvic aciduria). Pediatrics. 1957;20(2):290-302.
- 13. Brumm V, Bilder D, Waisbren S. Psychiatric symptoms and disorders in phenylketonuria. Molecular genetics and metabolism. 2010;99:S59-S63.
- 14. Abbaskhanian AZ, D; Afshar, P; Asadpoor, E; Rouhanizadeh, H; Jafarnia, A; Shokzadeh, M. Incidence of Neonatal Hyperphenylalaninemia Based on High-performance Liquid Chromatography Confirmatory Technique inMazandaran Province, Northern Iran (2007–2015). International Journal of Preventive Medicine. 2017;8(1):93.
- 15. Farhud D, Kabiri M. Incidence of phenylketonuria (PKU) in Iran. Indian journal of pediatrics. 1982;49(5):685-8.
- 16. Habib A, Fallahzadeh, M. H, Kazeroni, H. R. Ganjkarimi, A. H. Incidence of phenylketonuria in Southern Iran. Iranian Journal of Medical Sciences. 2015;35(2):137-9.
- 17. Motamedi N, Goodarzi E, Pordanjani SR, Valizadeh R, Moradi Y, Sohrabivafa M, et al. Incidence of phenylketonuriain Lorestan province, West of Iran (2006-2016). INTERNATIONAL JOURNAL OF PEDIATRICS-MASHHAD. 2017;5:4713-21.
- 18. Aoki K, Wada Y. Outcome of the patients detected by newborn screening in Japan. Pediatrics International. 1988;30(4):429-34.
- 19. Hanley W. Phenylketonuria (PKU)-What Next? Mini-Review. Journal of Genetic Disorders & Genetic Reports. 2016;2013.
- 20. Gu X, Wang Z, Ye J, Han L, Qiu W. Newborn screening in China: phenylketonuria, congenital hypothyroidism and expanded screening. Ann Acad Med Singapore. 2008;37(12 Suppl):107-4.
- 21. Berry SA, Brown C, Grant M, Greene CL, Jurecki E, Koch J, et al. Newborn screening 50 years later: access issues faced by adults with PKU. GeNetics in mediciNe. 2013;15(8):591.
- 22. Mathias D, Bickel H. Follow-up study of 16 years neonatal screening for inborn errors of metabolism in West Germany. European journal of pediatrics. 1986;145(4):310-2.
- 23. Zschocke J, Mallory J, Eiken HG, Nevin NC. Phenylketonuria and the peoples of Northern Ireland. Human genetics. 1997;100(2):189-94.
- 24. Özalp I, Coşkun T, Ceyhan M, Tokol S, Oran O, Erdem G, et al. Incidence of phenylketonuria and hyperphenylalaninaemia in a sample of the Turkish newborn population. Practical Developments in Inherited Metabolic Disease: DNA Analysis, Phenylketonuria and Screening for Congenital Adrenal Hyperplasia: Springer; 1986. p. 237-9.
- 25. Blau N. Genetics of phenylketonuria: then and now. Human mutation. 2016;37(6):508-15.
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32(3):338-43.
- 27. Farhud D, Shalileh M. Phenylketonuria and its dietary therapy in children. Iranian Journal of Pediatrics. 2008;18(Suppl 1):88-98.
- 28. Spécola N, Chiesa A. Alternative Therapiesfor PKU. Journal of Inborn Errors of Metabolism and Screening. 2017;5:2326409816685734.
اپیدمیولوژی تغذیه و سرطان: داستان دو شهر
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام