|
 دکتر محمدرضا ملک، phd بیوشیمی ،مشاور و مدرس سیستم کیفیت
دکتر محمدرضا ملک، phd بیوشیمی ،مشاور و مدرس سیستم کیفیتعارضه فسفولیپیدوز
عارضه فسفولیپیدوز یک اختلال در فرآیند ذخیرهسازی لیپیدها است که در آن مازاد فسفولیپیدها در بسیاری از سلولها از جمله هپاتوسیتها، لنفوسیتها و ماکروفاژها تجمع یافته و منجر به تغییرات ساختاری و عملکردی اندامکهای درونسلولی میشود. این عارضه نخستین بار توسط نلسون و فیتزوگ در سال 1948 با مشاهده ماکروفاژهای کفآلود در مدل حیوانی رت و به دنبال مصرف طولانیمدت داروی کلروکین گزارش شد. تغییرات مورفولوژیکی و فراساختاری اندامکها و سلولهای محیطی در حال حاضر بهعنوان یک نشانه مهم و حائز اهمیت جهت تشخیص عارضه فسفولیپیدوز است (شکل 1).
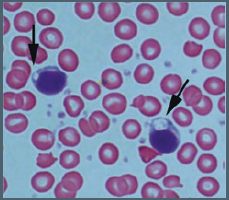
شکل 1: نمایی از تجمعات غیرطبیعی عارضه فسفولیپیدوز در لنفوسیتهای خون محیطی
به دنبال عارضه فسفولیپیدوز ناشی از مصرف داروهای کاتیونیک، بسیاری از مکانیسمهای فیزیکوشیمیایی در سلول مختل میشود. افزایش فعالیت ماکروفاژها، القای استرس اکسیداتیو، افزایش رادیکالهای آزاد، اثر بر پاسخ سیستم ایمنی هومورال و سلولار، اختلال در زنجیره انتقال الکترون میتوکندریال، کاهش فرآیند اتوفاژی و افزایش آپوپتوز از جمله این موارد هستند.
از جمله داروهایی که مصرف طولانیمدت آنها باعث بروز عارضه فسفولیپیدوز میشوند، میتوان به داروهای ضد افسردگی، ضد آنژین، ضد مالاریا، عوامل پایینآورنده کلسترول و برخی از آنتیبیوتیکها اشاره داشت. عارضه فسفولیپیدوز ناشی از داروهای کاتیونیک، با توجه به فراوانی مصرف این قبیل داروها، بخصوص در بیماریهایی که درمان آنها نیاز به مصرف طولانیمدت دارو توسط بیمار را دارد، امروزه تبدیل به یک نگرانی و چالش بزرگ برای صنایع داروسازی جهان جهت حفظ سلامت بیماران شده است. بر همین اساس بسیاری از تحقیقات با رویکرد شناخت بیشتر مکانیسمها و عوامل مداخلهگر در ایجاد عارضه فسفولیپیدوز ناشی از دارو در جهت بهبود اهداف درمانی با اثرات جانبی کمتر و کارایی بیشتر برای داروهای مصرفی طراحی میگردند. یک نکته کلیدی در کنار شناسایی هرچه بیشتر مکانیسمهای مختلف فسفولیپیدوز دارویی، پاسخ به این سؤال است که آیا بروز این عارضه میتواند منجر به سمیت سلولی شود یا اینکه تنها واکنشی خوشخیم به مصرف طولانیمدت داروهای کاتیونیک است. پاسخ به این سؤال هنوز دارای ابهامات زیادی است که خود توجیهی بر اهمیت این موضوع و انجام تحقیقات بیشتر در حال و آینده خواهد بود.
ساختار و مکانیسم عمل داروهای کاتیونیک
داروهای کاتیونیک ترکیباتی هستند که در ساختارشان یک یا چند حلقه هیدروفوب و یک زنجیره آمینی هیدروفیل وجود دارد (شکل 2). به دلیل داشتن این بخشهای هیدروفوبیک و هیدروفیلیک، توانایی عبور از غشاءهای بیولوژیکی از جمله سد خونی مغزی را دارا هستند، همچنین این ترکیبات بازهای ضعیف بوده و قادرند در شرایط اسیدی پروتونه شوند. ﮔﺮوه آﻣﯿﻦ زنجیره هیدروفیل این ترکیبات ﻗﺎﺑﻞ ﭘﺮوﺗﻮنه شدن بوده و ﺧﺎﺻﯿﺖ ﻟﯿﭙﻮﻓﯿﻠﯿﮏ دارد و ﻣﯽﺗﻮاﻧﺪ در اندامکهای سلولی مانند ﻟﯿﺰوزومﻫـﺎ یا ﻣﯿﺘﻮﮐﻨﺪريﻫﺎ ﺗﺠﻤﻊ ﯾﺎﺑﺪ. این تجمعات با اثرات زیانبار بر عملکرد این اندامکها همراه است و در نهایت منجر به انواع اختلالات متابولیکی در بدن میشود. تجمع داروهای کاتیونیک در اندامک لیزوزوم از ﻃﺮﯾـﻖ مکانیسم به دام افتادن pH اﺗﻔﺎق ﻣﯽاﻓﺘﺪ. این ترکیبات که در pH خنثی بدون بار الکتریکی هستند به محض ورود به ﻣﺤﯿﻂ اﺳﯿﺪي داﺧﻞ ﻟﯿﺰوزوم ﮔﺮوه آﻣـﯿﻦ آنها ﭘﺮوﺗﻮنه شده و در محیط لیزوزوم ﺑﻪ دام ﻣﯽاﻓﺘﺪ و در نتیجه ﻧﻤﯽﺗﻮاﻧﺪ دﯾﮕﺮ از اﻧﺪاﻣﮏ ﺧـﺎرج ﺷـﻮد. این فرآیند ﺑﺎﻋﺚ ﻣﻬﺎر آنزیمهای ﻓﺴﻔﻮﻟﯿﭙﺎز ﻟﯿﺰوزوﻣﯽ شده و در نهایت روند تخریب و تجزیه فسفولیپیدها مختل میشود و متعاقب آن تجمعات غیرطبیعی شامل دارو و فسفولیپیدها در درون اندامک لیزوزوم افزایش مییابند (شکل 3).
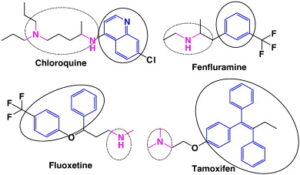
شکل 2: ساختار داروهای کاتیونیک

شکل 3: مکانیسم به دام افتادن داروهای کاتیونیک در اندامک لیزوزوم و القاء عارضه فسفولیپیدوز
فرآیند تشکیل تجمعات غیرطبیعی در اندامک ﻣﯿﺘﻮﮐﻨﺪري با مکانیسم دیگری انجام میشود. در این اندامک گروه آمین داروهای کاتیونیک در ﻓﻀﺎي ﺑﯿﻦ ﻏﺸﺎي داﺧﻠﯽ و ﺧﺎرﺟﯽ ﻣﯿﺘﻮﮐﻨﺪري ﭘﺮوﺗﻮنه شده و توسط نیروی پتانسیل منفی قوی به درون ﻓﻀﺎي ﻣﺎﺗﺮﯾﮑﺲ ﻣﯿﺘﻮﮐﻨﺪري کشیده میشود. این موضوع باعث اختلال در فرآیند اﮐﺴﯿﺪاﺳﯿﻮن و ﻓﺴﻔﺮﯾﻼﺳﯿﻮن اﮐﺴﯿﺪاﺗﯿﻮ شده و به دنبال آن تجمعات غیرطبیعی، اختلالات متابولیکی و تغییرات ساختاری شامل اﺳﺘﺌﺎﺗﻮز ﻣﯿﮑﺮووزﯾﮑﻮﻻر و سایر تغییرات در اندامکها به شکل آسیبهای جدی مانند نارسایی کبدی، کلیوی و قلبی بروز مینماید.
تغییرات ساختاری اندامکهای سلولی در عارضه فسفولیپیدوز ناشی از دارو
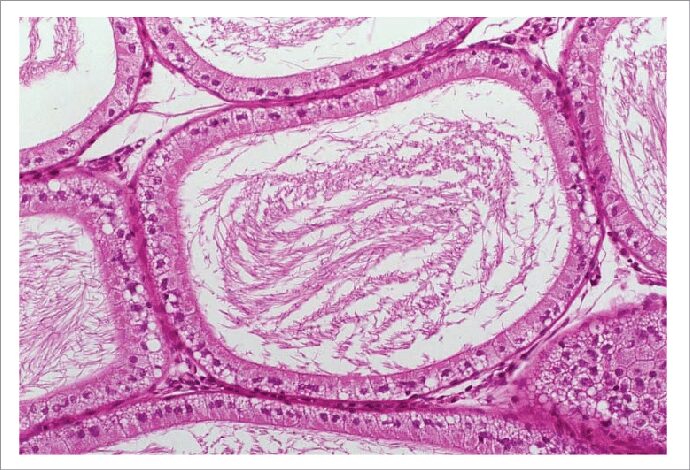
یکی از ویژگیهای فسفولیپیدوز، ایجاد تغییرات مورفولوژیکی در سلولهای تحت تأثیر است که به دنبال مصرف داروهای کاتیونیک که دارای پتانسیل بالا برای بروز این عارضه هستند در بافتهای مختلف ایجاد میشود. به کمک روش استاندارد طلایی میکروسکپ الکترونی عبوری میتوان تجمعات غیرطبیعی سلول به نام اجسام میلوئیدی یا اجسام لاملار را که از طریق به دام افتادن یا جذب انتخابی داروهای کاتیونیک درون لیزوزومها همراه با تجمع تدریجی آنها و افزایش مواد هضم نشده است را شناسایی نمود. لاملار بادیها بهعنوان یکی از مهمترین نشانههای اختصاصی جهت تشخیص عارضه فسفولیپیدوز دارویی کاربرد دارند. به دنبال افزایش چربیها و اکسیداسیون آنها و تولید رادیکالهای آزاد، واکنشهای التهابی ایجاد شده و ماکروفاژها را بیشتر فعال میکند. سلولهای ماکروفاژ چربیهای اکسیده را میبلعند و سیتوپلاسم آنها از قطرات حاوی چربی پر شده و به آنها ظاهر حبابدار و کفآلود میدهد (شکل 4). علاوه بر این، سایر تغییرات ساختاری و مورفولوژیکی نیز در سلولهای تحت اثر عارضه فسفولیپیدوز قابل شناسایی با استفاده از میکروسکپ الکترونی عبوری هستند؛ از جمله این تغییرات مورفولوژیکی میتوان به ایجاد ماکروفاژهای کفآلود، واکوئوله شدن سلول، تغییرات ساختاری در شبکه اندوپلاسمی، لیزوزومها و میتوکندریها و همچنین ایجاد استئاتوز سلولی اشاره کرد (شکل 5).
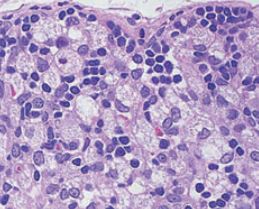
شکل 4: ماکروفاژها یا لنفوسیتهای کفآلود یا فومی شکل در عارضه فسفولیپیدوز
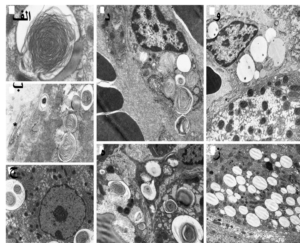
شکل 5: تغییرات مورفولوژیکی در بافت کبد ناشی از عارضه فسفولیپیدوز دارویی
الف) تشکیل لاملار بادی در لیزوزوم سلول هپاتوسیت که به علت عارضه فسفولیپیدوز ناشی از دارو ایجاد شده است. ب) ادغام دو لاملار بادی در سلول هپاتوسیت که همراه با مشاهده فیبرهای کلاژن در فضای سلولی است. ج) هیپرپلازی شبکه رتیکولوم اندوپلاسمیک که به علت عارضه فسفولیپیدوز دارویی در بافت کبد ایجاد شده است. د) نمایی از لاملار بادیها که بهوسیله سلولهای کوپفر در فضای سینوزوئیدال فاگوسیتوز شدهاند. ه) تجمع تعداد زیادی لاملار بادی در فضای داخلی هپاتوسیت که نشان از درجه بالای واکوئوله شدن سلول دارد. و) یک سلول کوپفر که از قطرات چربی پر شده است و یکی از مشخصههای مهم در عارضه فسفولیپدوز سلولی است. ز) قطرات چربی تشکیلشده در عارضه فسفولیپیدوز که نشاندهنده استئاتوز سلولی است.
عارضه فسفولیپیدوز در بافت کبد
با توجه به نقش مرکزی کبد در متابولیسم ترکیبات مختلف و نیز خنثی نمودن سمیت مواد و تسهیل دفع آنها، هرگونه اختلال و آسیب در این بافت میتواند باعث اثرات مضری بر فعالیتهای عملکردی آن بهخصوص سمزدایی داروها داشته باشد. به دنبال این اختلالات، مواد مختلف از جمله داروها در کبد تجمع یافته و آن را در مسیر آسیبهای شدید و مزمن قرار میدهند. بر همین اساس و با توجه به تنوع گروههای دارویی که مصرف بیرویه آنها میتواند باعث آسیبهای جدی به بافت کبد شود، شناسایی دقیق مکانیسمها و پاتوژنز داروهایی که بالقوه برای کبد سمی هستند، جهت اهداف درمانی با اثرات جانبی کمتر ارزشمند خواهد بود. همچنین با شناسایی بهموقع عارضه فسفولیپیدوز میتوان از بروز بیماریهای مزمن و خطرناک در بافت کبد که معمولاً در مراحل ابتدایی فاقد ویژگیهای بالینی اختصاصی هستند ولی با توسعه بیماری، شدت آسیبهای بافتی به شکل برگشتناپذیر افزایش خواهند یافت، جلوگیری نمود. بهعنوان مثال یکی از شایعترین آسیبهای مزمن کبدی، کبد چرب غیر الکلی است که در حالت پیشرفته تحت عنوان استئاتوهپاتیت غیر الکلی نامیده میشود و در صورت عدم تشخیص و درمان بهموقع در نهایت به سیروز کبدی و هپاتوسلولار کارسینوما تبدیل خواهد شد. از نظر بافتشناسی، کبد چرب به سه حالت ماکرووزیکولار، میکرووزیکولار و فسفولیپیدوز طبقهبندی میشود. اختلال در متابولیسم چربیها و تجمع آنها در کنار انباشت دارویی از جمله عوامل مهم در بروز التهاب و آسیبهای بافتی است که میتواند به دنبال فسفولیپیدوز دارویی ایجاد شود، هرچند که احتمال وقوع و مکانیسم آن بهدرستی شناخته نشده است. پنج مکانیسم عمده در عارضه فسفولیپیدوز باعث بروز آسیب به بافت کبد میشوند که از جمله آنها میتوان به اختلالات میتوکندریایی، متابولیتهای واکنشی و ایجاد نکروز سلولی، اختلالات لیزوزومال، مهار جریان صفراوی و ایجاد کلستاز و درگیری سیستم ایمنی اشاره نمود (شکل 6).
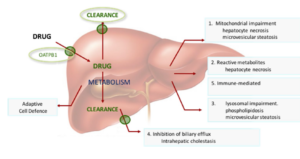
شکل 6: مکانیسمهای آسیب هپاتوسلولار در فسفولیپیدوز دارویی
مکانیسم بروز عارضه فسفولیپیدوز
مکانیسمهای مختلفی در بروز عارضه فسفولیپیدوز ناشی از مصرف طولانیمدت داروهای کاتیونیک مطرح است. بهطور کلی دو فرضیه، مکانیسم ایجاد عارضه فسفولیپیدوز را توضیح میدهد؛ فرضیه نخست بیان میکند که داروهای کاتیونیک مستقیماً به فسفولیپیدها متصل میشوند و در نتیجه کمپلکسهای غیر قابل تجزیه ایجاد میکنند که تجمع یافته و به شکل اجسام لاملار در اندامکهای لیزوزوم ذخیره میشوند. فرضیه دوم بر اساس مشاهداتی است که تولید لاملار بادیها را با مهار فعالیت آنزیمهای فسفولیپاز مرتبط میداند که یا به علت مهار مستقیم آنزیم و یا به دلیل برهمکنش داروهای کاتیونیک با دو لایه غشایی فسفولیپیدی لیزوزوم است؛ بهعنوان مثال آنزیم فسفولیپاز A2 به فسفولیپیدهای آنیونی در حضور غشاء لیزوزومال متصل شده و باعث تخریب فسفولیپیدها میشود. مطابق این تئوری در زمانی که داروهای کاتیونیک به غشاء لیزوزوم وصل میشوند، شارژ منفی غشاء کاهش یافته و به اتصال بین آنزیم و سوبسترا آسیب میزند و در نتیجه کاهش تخریب فسفولیپیدها اتفاق میافتد (شکل 7). مکانیسم فرضی دیگر برای ایجاد عارضه فسفولیپیدوز دارویی، رویکرد توکسیکوژنومیک این عارضه است که از طریق تنظیمات منفی و مثبت ژنهای مرتبط با بیوسنتز کلسترول و فسفولیپیدها، فعالیت فسفولیپازهای لیزوزومال و انتقال آنزیمها است.
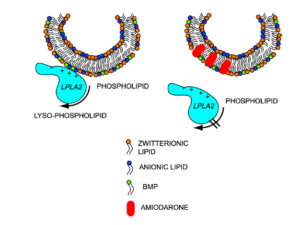
شکل 7: مدل پیشنهادی مهار آنزیم فسفولیپاز A2 لیزوزومال و القاء فسفولیپیدوز ناشی از داروی آمیودارون
روشهای تشخیصی عارضه فسفولیپیدوز ناشی از دارو
رویکردهای متفاوتی جهت مقاصد تشخیصی عارضه فسفولیپیدوز وجود دارد. مطالعات میکروسکپ الکترونی بهعنوان روش مرجع و استاندارد طلایی جهت بررسی حضور لاملار بادیها در بافت و سلولهای خون و بررسی سایر تغییرات مورفولوژیکی انجام میشود. از طرفی بافت کبد نسبت به عوامل مختلف آسیبرسان پاسخهای نسبتاً یکسانی میدهد. یکی از این پاسخها، تجمع غیرطبیعی ترکیبات مختلف و یا تغییراتی است که در نسبت تخریب ماتریکس خارج سلولی به سنتز آن اتفاق میافتد و معمولاً همراه با افزایش کلاژن بعلاوه سایر اجزاء ماده زمینهای خارج سلولی است. بر همین اساس، سنجش بیومارکرهای مرتبط با فرآیند فیبروزایی در بافت کبد مانند پروپپتید پروکلاژن تیپ سه، مهارکننده متالوپروتئیناز و هیالورونیک اسید با استفاده از روشهای ایمونواسی کمیلومینسانس یا الکتروکمیلومینسانس انجام میشود.
یکی دیگر از رویکردهای تشخیصی، انجام مطالعات متابونومیکس است. در این روش از اندازهگیری متابولیتهای اختصاصی مانند لیزو بیس فسفاتیدیک اسید یا بیس منو آسیل گلیسرو فسفات، فنیل استیل گلایسین و هیپوریک اسید در مایعات بدن مانند خون و ادرار استفاده میشود. این متابولیتها که مقدارشان در واکنش به مصرف داروهای کاتیونیک در مدلهای انسانی و حیوانی تغییر میکنند، با استفاده از تکنیکهای طیفسنجی رزونانس مغناطیسی هسته (NMR) و ﮐﺮوﻣﺎﺗﻮﮔﺮاﻓﻰ ﻣﺎﯾﻊ- طیفسنجی ﺟﺮمی (LC/MS) قابل اندازهگیری هستند؛ همچنین اندازهگیری سایر فسفولیپیدها مانند آسیل فسفاتیدیل گلیسرول، فسفاتیدیل کولین و فسفاتیدیل اینوزیتول میتواند به تشخیص کمک نماید. طبق تحقیقات انجامشده در عارضه فسفولیپیدوز ناشی از مصرف داروهای کاتیونیک، در مدل حیوانی رت، میزان فنیل استیل گلایسین افزایش و میزان هیپوریک اسید کاهش مییابد. افزایش نسبت فنیل استیل گلایسین به هیپوریک اسید شاخص قابل اعتمادی جهت مقاصد تشخیصی در عارضه فسفولیپیدوز ناشی از دارو است. تحقیقات نشان میدهد که تغییرات این دو متابولیت در مایعات خون و ادرار رت از همبستگی خوبی برخوردار است. مکانیسم افزایش نسبت فنیل استیل گلایسین به هیپوریک اسید به دلیل تغییر مسیر متابولیکی فنیل آلانین به سمت تولید بیشتر فنیل استیل گلایسین بهجای هیپوریک اسید بوده که این تغییر جهت به احتمال بسیار زیاد به علت مهار مسیر بتا اکسیداسیون است (شکل 8)
شکل 8: فرآیند تولید فنیل استیل گلایسین و هیپوریک اسید در عارضه فسفولیپیدوز دارویی
سنجش فعالیت آنزیمهای فسفولیپاز لیزوزومال، بررسی بیان ژنهای مرتبط، سنجش فسفولیپیدها در بافت و مایعات بدن و کشت سلولی از جمله سایر رویکردهای تشخیصی در عارضه فسفولیپیدوز ناشی از دارو هستند.
بهعنوان کلام آخر، باید توجه داشت که علیرغم تحقیقات مختلفی که در زمینه عارضه فسفولیپیدوز دارویی تاکنون به انجام رسیده است، هنوز ابهامات زیادی در این مورد وجود دارد و امید است تا با انجام تحقیقات بیشتر در آینده توسط دانشجویان دورههای تحصیلات تکمیلی و سایر محققان، شاهد شناسایی کاملتر و دقیقتر جنبههای مختلف تشخیصی و درمانی عارضه فسفولیپیدوز ناشی از دارو باشیم.
Reference:
- Nonoyama, T., Fukuda, R.)2008(. Drug-induced phospholipidosis.Pathological aspects and its prediction.J Toxical Pathol, 21, 9–24.
- Nelson, A., Fitzhugh, O. (1948). Chloroquine: Pathological changes observed in rats which for two years had been fed various proportions. Arch Pathol, 45, 454-462.
- Hruban, Z. (1976). Plumonary changes induced by amphiphilic drugs. Environ Health Perspect, 16, 111–118.
- Robison, R.L., Visscher, G.E., Roberts, S.A., Engstrom, R.G., Hartman, H.A., and Ballard, F.H. (1985). Generalized phospholipidosis induced by an amphiphilic cationic psychotropic drug. Toxicol Pathol, 13, 335–348.
- Lüllmann, H., Lüllmann-Rauch, R., and Wassermann, O. (1975). Drug induced phospholipidosis. Crit Rev Toxicol, 4, 185–218.
- Halliwell, W.H. (1997). Cationic Amphiphilic Drug-Induced Phospholipidosis.Toxicol Pathol, 25, 53-60.
- Reasor, M.J., Kacew, S. (2001). Drug-Induced Phospholipidosis: Are There Functional
- Anderson, N., Borlak, J., (2006). Drug-induced phospholipidosis. FEBS Letters, 580, 5533-5540.
- Monteith, D.K., Morgan, R.E., and Halstead, B. (2006). In vitro assays and biomarkers for drug-induced phospholipidosis. Expert Opin Drug Metab Toxicol, 2, 687-696.
- 10. Reasor, M.J. (1989). A review of the biology and toxicology implications of the induced of lysosomal lamellar bodies by drugs. Toxicol Appl Pharmacol, 97, 47–56.
- Kodavanti, U.P., and Mehendale, H.M. (1990). Cationic amphiphilic drugs and phospholipid storage disorder. Pharm Rev, 42, 327–353.
- Reasor MJ. Phospholipidosis in the alveolar macrophage induced by cationic amphiphilic drugs. Fed Proc. 43: 2578– 2581. 1984.
- Edinger, A.L., and Thompson, C.B. (2004). Death by design: apotosis, necrosis and autophagy. Curr Opin Cell Biol, 16, 663-669.
- Sarma, J.S., Pei, H., and Venkataraman, K. (1997). Role of oxidative stress in amiodarone-induced toxicity. J Cardiovasc Pharmacol Ther, 2, 53–60.
- Mingeot-Leclercq, M.P., and Tulkens, P.M. (1999). Aminoglycosides: Nephrotoxicity. Antmicrob Agents Chemother, 43, 1003– 1012.
- Mortuza, G.B., Neville, W.A., Delaney, J., Waterfield, C.J., and Camilleri, P. (2003). Characterization of a potential biomarker of phospholipidosis from amiodarone-treated rats. Biochem Biophys Acta, 1631, 136–146.
- Hung, D.Y., Chang, P., Cheung, K., McWhinney, B., Masci, P.P., Weiss, M., Roberts, M.S. (2002). Cationic drug pharmacokinetics in diseased livers determined by fibrosis index, hepatic protein content, microsomal activity, and nature of drug. J Pharmacol Exp Ther, 301(3), 1079-87.
- Natalie, M., Margino, S., Erik, H., Geert, V., Freddy, V.G., Jaques, V.G. (2010). Screening for phospholipidosis induced by central nervous drugs: Comparing the predictivity of an in vitro assay to high throughput in silico assays. Toxicology in vitro, 1417–1425.
- Raddatz, D., Ramadori, G. (2007). Carbohydrate metabolism and the liver: actual aspects from physiology and disease. Z Gastroenterol, 45(1), 51-62.
- Dragovic, S., Vermeulen, N., Gerets, H., Hewitt, P., Ingelman‐Sundberg, M., Park, B. K., Juhila, S., Snoeys, J., Weaver, R.J. (2016). Evidence‑based selection of training compounds for use in the mechanism‑based integrated prediction of drug‑induced liver injury in man. Arch Toxicol, 90, 2979–3003.
- Canbay, A., Bechmann, L., Gerken, G. (2007). Lipid metabolism in the liver. Z Gastroenterol, 45(1), 35-41.
- Wong, J.B., McQuillan, G.M., McHutchison, J.G., Poynard, T. (2000). Estimating Future Hepatitis C Morbidity, Mortality, and Costs in the United States. Am J Public Health, 90(10), 1562–1569.
- Brenner, D.A. (2009). Molecular Pathogenesis of Liver Fibrosis. Trans Am Clin Climatol Assoc, 120, 361–368.
- Paschos, P., and Paletas, K. (2009). Non alcoholic fatty liver disease and metabolic syndrome.Hippokratia, 13(1), 9–19.
- Shin, Y.H., Ko, J.S., Kim, G.S., Gwak, M.S., Sim, W.S., Lee, A.R., Yi, H.W., Joh, J.W. (2012). Impact of hepatic macrovesicular and microvesicular steatosis on the postoperative liver functions after right hepatectomy in living donors. Transplant Proc, 44(2), 512-5.
- Brandao, D.F., Ramalho, L.N., Ramalho, F.S., Zucoloto, S., Martinelli, A. L., Silva ,O. C. (2006). Liver cirrhosis and hepatic stellate cells. Acta Cir Bras, 1, 54-7.
- Tsuchida, T., Friedman, S.L. (2017). Mechanisms of hepatic stellate cell activation.Nat Rev Gastroenterol Hepatol, 14(7), 397-411.
- Faty, A., Ferre, P., Commans, S. (2012). The acute phase protein Serum Amyloid A induces lipolysis and inflammation in human adipocytes through distinct pathways. PLoS One, 7(4), e34031.
29. Itoh, S., and Tsukada, Y. (1973). Clinico-pathological and electron microscopical studies on a coronary dilating agent: 4, 4′-diethylaminoethoxyhexesterol-induced liver injuries. Acta Hepato-Gastroent, 20, 204-215 - Sawada, H., Takami, K., Asahi, S. (2005). A toxicogenomic approach to drug-induced phospholipidosis: Analysis of its induction mechanism and establishment of a novel in vitro screening system. Toxicol Sci, 83, 282-292.
- Abe, A., Shayman, J.A. (2009). The role of negatively charged lipids in lysosomal phospholipase A2 function. J. Lipid Res, 50, 2027-2035.
- Delaney, J., Neville, W.A., Swain, A., Miles, A., Leonard, M.S., and Waterfield, C.J. (2004). Phenylacetyl glycine, a putative biomarker of phospholipidosis: its origins and relevance to phospholipid accumulation using amiodarone treated rats as a model. Biomarkers, 9, 271-290.
- Shayman, J.A., Abe, A. (2013). Drug-induced phospholipidosis: An acquired lysosomal storage disorder. Biochim Biophys Acta, 1831, 602-611.
- Rahimi, R.S., Rockey, D.C. (2011). Complications and outcomes in chronic liver disease. Curr Opin Gastroenterol, 27(3), 204-9.
- Schuppan, D., and Nezam, H. (2008). Afdhal. Liver Cirrhosis. Lancet. 8, 371(9615), 838–851.
- Crockett, S.D., Kaltenbach, T., Keeffe, E.B. (2006). Do We Still Need a Liver Biopsy? Are the Serum Fibrosis Tests Ready for Prime Time? Clin Liver Dis, 10, 514–534.
- Manning, D.S., Afdhal, N.H. (2008). Diagnosis and Quantitation of Fibrosis. Gastroenterology, 134, 1670–1681.
- Rosenberg, W., Voelker, M., Thiel, R., Becka, M., Burt, A., Schuppan, D., Hubscher, S., Toskams, T., Pinzani, M., Arthur, M. (2004). On behalf of the European Liver Fibrosis Group. Serum markers detect the presence of liver fibrosis: A cohort study.Gastroenterology, 127, 1704–1713.
- Liu, N., Tengstrand, E.A., Chourb, L., Hsieh, F.Y. (2014). Di-22:6-bis (monoacylglycerol)phosphate: A clinical biomarker of drug-induced phospholipidosis for drug development and safety assessment. Toxicol Appl Pharmacol, 279(3), 467-76.
- Kamiguchi, H., Murabayashi, M., Mori, I., Horinouchi, A., Higaki, K. (2016). Biomarker discovery for drug-induced phospholipidosis: phenylacetylglycine to hippuric acid ratio in urine and plasma as potential. Biomarkers, 22, 178-188
نقش سیستم اپیوئیدی اندوژن در فسفولیپیدوز ناشی از کلروکین در کبد رت
بررسی میزان استرس اکسیداتیو سرم و مایع آسیت بیماران مبتلا به سیروز کبدی
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام