تشخیص آزمایشگاهی و درمان کمبود توأم فاکتورهای انعقادی
1- کمبود مرکب فاکتورهای انعقادی، تشخیص و درمان (ادامه)
2- کمبود فاکتورهای انعقادی وابسته به ویتامین K
اکبر درگلاله1، حسن مروتی2
1- گروه هماتولوژی و طب انتقال خون، دانشگاه علوم پزشکی ایران
2- عضو هیئت علمی مرکز تحقیقات واکسن و سرمسازی رازی
تشخیص کمبود مشترک فاکتور V و فاکتور VIII
فنوتیپ خونریزی F5F8D، نسبتاً مشابه با تصویر بالینی کمبود مجزای فاکتور V و فاکتور VIII است، بنابراین برخی موارد ممکن است به اشتباه هموفیلی A خفیف یا کمبود فاکتور V تشخیص داده شوند و نقایص یکی از فاکتورها ممکن است نادیده گرفته شود (4). چنین خطاهایی نشان میدهند که ممکن است در تعداد دقیق بیماران مبتلا به F5F8D، در برخی کشورها یک عدم صحت وجود داشته باشد؛ به ویژه در جاهایی که آزمونهای آزمایشگاهی و ژنتیکی ضعیفی وجود دارد.
حضور همزمان کمبود فاکتور V چه با هموفیلی A و چه VWD، تشخیص افتراقی اصلی F5F8D است. با اتکا به آزمایشهای اختصاصی VWD میتوان F5F8D را از وراثت همزمان کمبود فاکتور V و VWD افتراق داد. افتراق F5F8D از وراثت همزمان کمبود فاکتور V و هموفیلی A عمدتاً بر اساس سابقه فامیلی است. ممکن است در F5F8D هیچ شواهدی از سابقه فامیلی مثبت وجود نداشته باشد، با این حال اگر هم وجود داشته باشد الگوی وراثت بهصورت اتوزوم مغلوب خواهد بود (24).
بهطور معمول به دنبال کاهش همزمان در سطوح پلاسمایی فاکتور V و فاکتور VIII؛ معمولاً بین 5% و 20%، به F5F8D شک میکنیم. بهطور کلی، طولانی شدن PT و aPTT همراه با زمان ترمبین [1](TT) نرمال، بیانگر کمبود فاکتورهای انعقادی مسیر مشترک است. تست میکسینگ (50:50) میتواند وجود مهارکنندهها را رد کند و کاهش فعالیت هر دو فاکتور V و VIII به IU/mL0/05–0/2 و بهندرت به کمتر از IU/mL0/05، این اختلال را تأیید میکند (1, 5). در نهایت، میتوان از آنالیز مولکولی ژنهای MCFD2 و LMAN1 برای تأیید F5F8D استفاده کرد، اگرچه این روش بهطور روتین استفاده نمیشود (21).
به دلیل فنوتیپ خونریزیدهنده خفیف F5F8D، تشخیص پیش از تولد معمولاً توصیه نمیشود. با این حال اگر تمایل به انجام آن وجود دارد، هر دو والدین باید حامل بیماری باشند؛ در صورتی که والدین در حال حاضر یک فرزند مبتلا داشته باشند میتوان به این مسئله پی برد. نمونهبرداری از پرزهای جفتی [2](CVS) باید در هفته 12-10 بارداری انجام شود. سپس DNA جنینی برای موتاسیونهای خاصی که در والدین و فرزند مبتلا یافت شده، بررسی میشود (25).
مدیریت
تمایل به خونریزی در کمبود مشترک، از کمبود مجزای فاکتور V یا فاکتور VIII شدیدتر نیست. تظاهرات بالینی معمولاً خفیف است، بنابراین عمدتاً درمان تقاضامحور[3] برای مدیریت خونریزی در اقدامات جراحی یا در زمان زایمان و در مواردی با وقایع خونریزیدهنده تروماتیک ترجیح داده میشود. در F5F8D، بهجز در موارد خاص با خونریزیهای مکرر و شدید، نیازی به پروفیلاکسی وجود ندارد (25).
هر دو فاکتور V و VIII فاکتورهای ناپایدار هستند و به ترتیب نیمهعمر کوتاهی حدود 36-16 ساعت و 14-10 دارند؛ بنابراین، پلاسمای تازه منجمد [4](FFP) معمولاً برای رساندن فعالیت فاکتور V به سطح هموستاتیک کفایت میکند، اما ممکن است برای فعالیت فاکتور VIII کافی نباشد.
درمان تقاضامحور عبارتست از: FFP فاقد ویروس[5] برای جبران فاکتور V، همراه با 1-دسآمینو-8دی-آرژنین وازوپرسین [6](DDAVP) در بیماران با کمبود خفیف فاکتور VIII، یا همراه با کنسانترههای فاکتور VIII مشتقشده از پلاسما/ نوترکیب در بیماران با کمبود متوسط/ شدید فاکتور VIII (جدول 5-9) (26).
برای جراحی، مقادیر هدف فاکتور V و فاکتور VIII در گردش، باید به ترتیب بیشتر از 20% و 50% باشد (26). برای خونریزیهای شدید یا جراحیهای بزرگ، توصیه میشود که ســـــطوح فاکتور V و فاکتور VIII با ml/kg 25-15 FFP مجاور شده با حلال-پاککننده [7](SD-FFP) جبران شود و جایگزینی بیشتر فاکتور VIII با IU/kg 20-40 کنسانتره فاکتور VIII نوترکیب [8](rFVIII) یا μg/kg 0/3 دسموپرسین صورت بگیرد. برای حفظ فعالیت فاکتور V و فاکتور VIII در مقادیر هدف برای جراحیهای بزرگ، ادامه درمان با فواصل 12 ساعته ضروری است (5). برای خونریزیهای خفیف و جراحیهای کوچک، ml/kg 20-15 ترنکسامیک اسیــد یا تنها g1، چهار بار در روز توصیه میشود (5).
در دوران بارداری، سطح فاکتور VIII بهطور پیشروندهای افزایش پیدا میکند، درحالیکه فاکتور V بدون تغییر باقی میماند؛ بنابراین معمولاً در زنان باردار مبتلا به F5F8D، فعالیت فاکتور VIII برای هموستاز کافی است، اما سطح هموستاتیک فاکتور V در زمان زایمان کافی نیست (27). زنان با F5F8D در معرض خطر خونریزی پس از زایمان هستند، بنابراین باید در طی بارداری و پس از آن مدیریت شوند. در زنان با فعالیت فاکتور V کمتر از 20% در سه ماهه سوم، تجویز یک دوز SD-FFP (ml/kg 15-25) در زمان زایمان یا قبل از عمل سزارین توصیه میشود. درمان بیشتر با SD-FFP با دوز ml/kg 10 در فواصل 12 ساعته برای حفظ فعالیت فاکتور V به بیشتر از 20% به مدت حداقل 72 ساعت باید مد نظر قرار بگیرد. در بیماران با فعالیت فاکتور VIII زیر 50% در سه ماهه سوم، rFVIII مازاد نیز توصیه میشود (5).
جدول (5-9) گزینههای درمانی مختلف برای مدیریت بیماران با کمبود مشترک فاکتور V و فاکتور VIII در شرایط مختلف
| موقعیت | دوزهای توصیهشده |
| خونریزیهای خفیف/ جراحیهای کوچک | ترنکسامیک اسید (ml/kg 15-20 یا g 1 چهار بار در روز) |
| خونریزیهای شدید/ جراحیهای بزرگ |
SD-FFP (ml/kg 15-25) + دسموپرسین (μg/kg0/3) (28) برای کمبود خفیف فاکتور VIII یا کنسانترههای rFVIII (IU/kg20-40) برای کمبود متوسط/ شدید فاکتور VIII |
| بارداری (فعالیت فاکتور V کمتر از 20% در سه ماهه سوم) |
SD-FFP (ml/kg15-25)، یک دوز در زمان زایمان یا قبل از عمل سزارین + SD-FFP (ml/kg 10) با فواصل 12 ساعته حداقل به مدت 3 روز اگر فعالیت فاکتور VIII کمتر از 50% در سه ماهه سوم باشد: + rFVIII اضافی
|
| SD-FFP: پلاسمای تازه منجمد مجاور شده با حلال- پاککننده،
rFVIII: فاکتور VIII نوترکیب |
|
کمبود فاکتورهای انعقادی وابسته به ویتامین K
پیشگفتار
کمبود فاکتورهای انعقادی وابسته به ویتامین K [9](VKCFD) یک اختلال خونریزیدهنده نادر است که تاکنون در کمتر از 50 خانواده گزارش شده است (29). مقالات، تنها محدود به تعداد کمی از گزارشهای موردی و گزارش مواردی با تعداد اندک بیماران است. این بیماری یک اختلال اتوزوم مغلوب است که ناشی از نقص ژنهای γ- گلوتامیل کربوکسیلاز [10](GGCX) یا زیرواحد 1 کمپلکس ویتامین K اپوکسید ردوکتاز [11](VKORC1) است. در صورتی که موتاسیون در آنزیم اول باشد بهعنوان اختلال تایپ I در نظر گرفته میشود و اگر موتاسیون عامل بیماری، آنزیم دوم را درگیر کند بهعنوان اختلال تایپ II در نظر گرفته میشود (30). این ژنها کدکننده پروتئینهایی هستند که در γ- کربوکسیلاسیون اسید آمینههای گلوتامات (Glu) موجود در فاکتورهای انعقادی وابسته به ویتامین K، شامل فاکتورهای II، VII، IX و X، نقش دارند. VKCFD معمولاً در نوزادی و با وقایع خونریزیدهنده تهدیدکننده حیات بروز مییابد (31). تشخیص VKCFD با طولانی شدن PT و aPTT، همراه با TT نرمال و کاهش توأم فاکتورهای انعقادی وابسته به ویتامین K، معمولاً حدود 1% تا 30%، امکانپذیر است. مدیریت بیماری عمدتاً از طریق تجویز ویتامین K1 یا کنسانتره کمپلکس پروترومبین [12](PCC) چهار- فاکتوری است (جدول 6 فصل 10) (5).
ساختار و عملکرد GGCX و VKOR
GGCX یک پروتئین سراسری ترانس ممبرن است که در غشای شبکه اندوپلاسمی (ER) قرار دارد. این پروتئین حاوی 758 اسید آمینه است و یک پیوند دیسولفید بین سیستئینهای 99 و 450 وجود دارد (31). اطلاعات محدودی در رابطه با ساختار GGCX موجود است، با این حال بهنظر میرسد که حاوی 5 دامین ترانس ممبرن است. انتهای آمینی این آنزیم در سیتوپلاسم قرار دارد و انتهای کربوکسیل در معرض لومن ER است (32). بر اساس مطالعاتی که بر روی ساختار و مکانیسم عمل GGCX صورت گرفته است، جایگاههای اتصالی و کاتالیتیک متفاوتی برای آن در نظر گرفته میشود. بر این اساس، GGCX دارای یک جایگاه اتصالی پروپپتید، یک جایگاه اتصالی گلوتامات، یک جایگاه اتصالی ویتامین K، یک جایگاه فعال کربوکسیلاسیون، یک جایگاه فعال اپوکسیداسیون و احتمالاً یک جایگاه اتصالی CO2 است؛ اما در رابطه با محل دقیق این نواحی عملکردی اطلاعات محدودی وجود دارد (32).
پروتئین VKOR نیز یک پروتئین سراسری در غشای ER با 163 اسید آمینه است (31). دو مدل توپولوژی برای این پروتئین وجود دارد که سه یا چهار دامین ترانس ممبرن را برای پروتئین متصور میشود. بر اساس مطالعات مختلف، مدل “سه دامین ترانس ممبرن”[13] منطقیتر است. در این مدل، انتهای آمینی VKOR در لومن ER و انتهای کربوکسیل آن در سیتوپلاسم قرار گرفته است (32). برای مدتهای طولانی تصور بر این بود که VKOR یک کمپلکس چند آنزیمی است؛ نظریهای که امروزه مورد سؤال قرار گرفته است (32).
چرخه ویتامین K
کربوکسیلاسیون وابسته به ویتامین K نوعی اصلاح پس از ترجمه[14] است که برای عملکرد صحیح پروتئینهای وابسته به ویتامین K ضروری است. مهمترین پروتئینهای وابسته به ویتامین K عبارتند از فاکتورهای انعقادی II، VII، IX و X، ضد انعقادهای طبیعی پروتئین C، پروتئین S و پروتئین Z، پروتئینهای استخوانی شامل استئوکلسین، پروتئین ماتریکس Gla و پروتئین اختصاصی توقف رشد-6 [15](Gas6) (33).
در کربوکسیلاسیون وابسته به ویتامین K، اسید آمینههای گلوتامات (Glu) خاصی به γ-کربوکسی گلوتامات (Gla) تغییر میکنند. هر فاکتور وابسته به ویتامین K حاوی 12-10 اسید آمینهی Gla در انتهای آمینی است که دامین Gla نامیده میشود. آنزیم مسئول این واکنش، GGCX است که به ویتامین K احیا (KH2)، CO2 و O2 بهعنوان کوفاکتور نیاز دارد. زمانی که هر Glu تغییر میکند، یک مولکول KH2 به ویتامین K 2، 3 اپوکسید (KO)، اکسید میشود. برای تجدید KH2، این مولکول KO باید دوباره به شکل احیاشده تبدیل شـود (شکل 4-9). این تبدیل در یک واکنش دو مرحلهای اتفاق میافتد؛ ابتدا KO با کمک VKOR به ویتامین K احیا میشود و سپس ویتامین K با کمک ویتامین K ردوکتاز به KH2 احیا میگردد (32, 34).
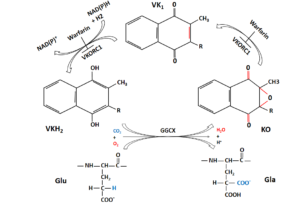
شکل (4-9) کربوکسیلاسیون گلوتامات (Glu) به اسید آمینههای کربوکسی گلوتامات (Gla) بهوسیله GGCX برای فعال شدن فاکتورهای انعقادی وابسته به ویتامین K ضروری است. VKH2 کوفاکتور این واکنش است. در این روند، VKH2 به KO اکسید میشود و سپس بهوسیله VKORC1 به ترتیب به ویتامین K کینون و VKH2 احیا میشود. وارفارین میتواند VKORC1 را مسدود کند. ردوکتاز دیگری به نام NQO1 نیز میتواند VK را به VKH2 تبدیل کند. وارفارین میتواند VKORC1 را مهار کند اما NQO1 را مهار نمیکند (35). GGCX: γ– گلوتامیل کربوکسیلاز، VKH2: ویتامین K هیدروکینون، KO: ویتامین K اپوکسید، VKORC1: زیرواحد 1 کمپلکس ویتامین K اپوکسید ردوکتاز، NQO1: NADPH کینون اکسیدو ردوکتاز، VK: ویتامین K
گاما کربوکسیلاسیون اسید آمینههای Glu برای اتصال یونهای کلسیم ضروری است که پس از آن امکان اتصال فاکتورهای وابسته به ویتامین K را به غشاهای فسفولیپیدی همچون سطح پلاکتهای فعال یا اندوتلیوم آسیبدیده فراهم میکند. این پدیده باعث تجمع فاکتورهای انعقادی در محل آسیب عروقی میشود (32).
کمبود فاکتورهای انعقادی وابسته به ویتامین K
VKCFD (OMIM: 277450# و 607473#) اولین بار در سال 1966 توسط McMillan و Robert در یک دختربچه 4 ماهه توصیف شد (36). علائم وی با کبودیها و وقایع خونریزیدهنده متعدد، PT و aPTT طولانی و سطوح غیر قابل شناسایی فاکتورهای II، VII، IX و X در آزمونهای انعقادی تظاهر یافته بود. با این حال، نه بیماری کبدی و نه سوءجذب شناسایی نشدند. سطوح پایین فاکتورهای انعقادی پس از تجویز دوزهای بالای ویتامین K یک بهبودی نسبی را نشان داد (36). بیمار در سن 15 سالگی تحت بررسیهای بیشتر قرار گرفت و فاکتورهای انعقادی مجدداً با آزمونهای ایمونولوژیک ارزیابی شدند (37) ولی مکانیسم مولکولی نامشخص باقی ماند.
امروزه VKCFD بهعنوان یک اختلال خونریزیدهنده اتوزوم مغلوب شناخته میشود که ناشی از نقایص ژن GGCX یا VKORC1 است. سطوح پلاسمایی فاکتورهای انعقادی وابسته به ویتامین K در VKCFD ممکن است حدود 1% تا 30% باشد (1). VKCFD تاکنون در کمتر از 50 خانواده گزارش شده است (1, 29).
تظاهرات بالینی
VKCFD معمولاً در نوزادی تظاهر مییابد، هرچند ممکن است برای مدت کوتاهی نهفته باقی بماند. شدت تظاهرات بالینی به سطح فاکتورهای انعقادی کاهشیافته بستگی دارد (6)، با این حال، تصویر بالینی با فعالیت فاکتورهای انعقادی وابسته به ویتامین K همبستگی نزدیکی ندارد (5). خونریزیهای شدید همچون خونریزی درون جمجمهای (ICH) یا خونریزی از بند ناف در نوزادان تازه متولد مبتلا توصیف شده است (38-40). خونریزی پوستی- مخاطی و بافت نرم و خونریزیهای پس از تروما نیز در این بیماران گزارش شده است (جدول 6-9) (38, 41). VKCFD ممکن است با شیوع کمتری با خونریزی در بزرگسالی خود را نشان دهد و یا حتی بهطور تصادفی شناسایی شود.
جدول (6-9) تظاهرات بالینی کمبود فاکتورهای انعقادی وابسته به ویتامین K
| تظاهرات بالینی | وقوع |
| خونریزی درون جمجمهای | 34% |
| اکیموز/ کبودی آسان | 21% |
| ناهنجاریهای اسکلتی/ تأخیر در رشد یا تکامل | 21% |
| خونریزی بند ناف | 17% |
| پس از تروما/ پس از جراحی | 17% |
| خونریزی بینی | 17% |
| لثه/ حفره دهان | 12% |
| همارتروز | 4% |
بعضی از افراد مبتلا ممکن است از عقبماندگی ذهنی و ناهنجاریهای اسکلـــــــــــتی نیز رنج ببرند که به γ- کربوکسیلاسیون معیوب سایر پروتئینهای وابسته به ویتامین K نسبت داده میشود (39, 42). ناهنجاریهای اسکلتی شامل هایپوپلازی بینی، هایپوپلازی دیستال دیژیتال[16] و منقوط شدن اپیفیز[17] مشابه با مواردی است که در امبریوپاتی وارفارین[18] دیده میشود (43). اختلالات مشابه سودوگزانتوم الاستیکوم نیز در بیماران مبتلا به VKCFD با موتاسیونهای GGCX گزارش شده است (44).
بعضی ضد انعقادهای طبیعی شامل پروتئین C، پروتئین S و پروتئین Z نیز نیازمند تبدیل اسید آمینههای Glu به اسید آمینههای γ- کربوکسی گلوتامات (Gla) هستند و بنابراین در نقایص GGCX و VKOR سطوح پروتئین C و پروتئین S نیز پایین است. این واقعیت که تاکنون هیچ موردی با ترومبوز در مقالات گزارش نشده است، ممکن است بیانگر اثر غالب این دو آنزیم در فعالیتهای پیشانعقادی باشد (29, 45).
اساس مولکولی
ژن کدکننده GGCX با طول 13 کیلوباز بر روی کروموزوم 2p11.2 واقع شده است و شامل 15 اگزون است. ژن کدکننده پروتئین VKORC1 که VKORC1 نامیده میشود بر روی کروموزوم 16p11.2 قرار گرفته است. VKORC1 یک ژن کوچک با طول 5126 جفت باز و شامل 3 اگزون است (6). نقص در GGCX بهعنوان VKCFD تایپ I شناخته میشود. آنزیم دیگری که در این چرخه نقش مهمی ایفا میکند، VKORC1 است. VKORC1 تبدیل دوباره ویتامین K اپوکسید (KO) را که طی آخرین واکنش تولید شده است، به KH2 کاتالیز میکند (شکل 4-9). نقص در VKORC1 بهعنوان VKCFD تایپ II شناخته میشود (5, 33).
تاکنون حداقل 18 موتاسیون در ژن GGCX گزارش شده است که با VKCFD مرتبط هستند و اکثر آنها موتاسیونهای نقطهای هستند (جدول 7-9) (6, 29, 46). به نظر میرسد یک موتاسیون بدمعنی که منجر به جایگزینی تریپتوفان با آرژنین در اسید آمینه شماره 98 میشود، تنها موتاسیون گزارششده است که VKORC1 را درگیر میکند (جدول 7-9).
اخیراً Jin و همکاران نشان دادند که 1657delA و IVS13-6G>A موتاسیونهای عامل بیماری در اولین بیمار VKCFD هستند که توسط McMillan و Robert گزارش شد (10).
منابع:
- Castaman G, Linari S. Diagnosis and treatment of von Willebrand disease and rare bleeding disorders. Journal of clinical medicine. 2017;6(4):45.
- Seligsohn U, Zivelin A, Zwang E. Combined factor V and factor VIII deficiency among non-Ashkenazi Jews. New England Journal of Medicine. 1982;307(19):1191-5.
- Peyvandi F, Tuddenham E, Akhtari A, Lak M, Mannucci P. Bleeding symptoms in 27 Iranian patients with the combined deficiency of factor V and factor VIII. British journal of haematology. 1998;100(4):773-6.
- Zhang B. Recent developments in the understanding of the combined deficiency of FV and FVIII. British journal of haematology. 2009;145(1):15-23.
- Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, et al. Guideline for the diagnosis and management of the rare coagulation disorders: a United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British Committee for Standards in Haematology. British journal of haematology. 2014;167(3):304-26.
- Zhang B, Ginsburg D. Familial multiple coagulation factor deficiencies: new biologic insight from rare genetic bleeding disorders. Journal of Thrombosis and Haemostasis. 2004;2(9):1564-72.
- Zheng C, Liu H-h, Yuan S, Zhou J, Zhang B. Molecular basis of LMAN1 in coordinating LMAN1-MCFD2 cargo receptor formation and ER-to-Golgi transport of FV/FVIII. Blood. 2010;116(25):5698-706.
- Zheng C, Liu H-h, Zhou J, Zhang B. EF-hand domains of MCFD2 mediate interactions with both LMAN1 and coagulation factor V or VIII. Blood. 2010;115(5):1081-7.
- Oeri J, Matter M, Isenschmid H, Hauser F, Koller F. Congenital factor V deficiency (parahemophilia) with true hemophilia in two brothers. Bibliotheca paediatrica. 1954;58:575.
- Jin D-Y, Ingram BO, Stafford DW, Tie J-K. Molecular basis of the first reported clinical case of congenital combined deficiency of coagulation factors. Blood. 2017;130(7):948-51.
- Nichols WC, Seligsohn U, Zivelin A, Terry VH, Arnold ND, Siemieniak DR, et al. Linkage of combined factors V and VIII deficiency to chromosome 18q by homozygosity mapping. The Journal of clinical investigation. 1997;99(4):596-601.
- Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, et al. Mutations in the ER–Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93(1):61-70.
- Shetty S, Madkaikar M, Nair S, Pawar A, Baindur S, Pathare A, et al. Combined factor V and VIII deficiency in Indian population. Haemophilia: the official journal of the World Federation of Hemophilia. 2000;6(5):504-7.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA: a cancer journal for clinicians. 2017;67(1):7-30.
- Viswabandya A, Baidya S, Nair S, Lakshmi K, Mathews V, George B, et al. Clinical manifestations of combined factor V and VIII deficiency: a series of 37 cases from
lacement therapy in a patient with combined factor V (FV) and FVIII deficiency due to novel homozygous mutation in LMAN1. Haemophilia. 2015;21(6):e492-e4.
- Wang A, Liu X, Wu J, Cai X, Zhu W, Sun Z. Combined FV and FVIII deficiency (F5F8D) in a Chinese family with a novel missense mutation in MCFD 2 gene. Haemophilia. 2014;20(6):e436-e8.
- Zheng C, Zhang B, editors. Combined deficiency of coagulation factors V and VIII: an update. Seminars in thrombosis and hemostasis; 2013: Thieme Medical Publishers. a single center in India. American journal of hematology. 2010;85(7):538-9.
- Elmahmoudi H, Wigren E, Laatiri A, Jlizi A, Elgaaied A, Gouider E, et al. Analysis of newly detected mutations in the MCFD2 gene giving rise to combined deficiency of coagulation factors V and VIII. Haemophilia. 2011;17(5):e923-e7.
- Hejer E, Adnen L, Asma J, Ibtihel M, Benammar-Elgaaied A, Gouider E. Identification of a novel mutation in the MCFD2 gene in a Tunisian family with combined factor V and VIII deficiency. La Tunisie medicale. 2012;90(4):343-4.
- Karimi M, Cairo A, Safarpour MM, Haghpanah S, Ekramzadeh M, Afrasiabi A, et al. Genotype and phenotype report on patients with combined deficiency of factor V and factor VIII in Iran. Blood coagulation & fibrinolysis. 2014;25(4):360-3.
- Wang A, Duan Q, Ding K, Liu X, Wu J, Sun Z. Successful abdominal operation without rep
- Genotypes of patients with combined factor V and VIII deficiency [internet]. 2011. https://c.ymcdn.com/sites/www.isth.org/resource/resmgr/publications/fv_and_viii_mutations-2011.pdf.
- Zhang B, Spreafico M, Zheng C, Yang A, Platzer P, Callaghan MU, et al. Genotype-phenotype correlation in combined deficiency of factor V and factor VIII. Blood. 2008;111(12):5592-600.
- Khoriaty R, Vasievich MP, Ginsburg D. The COPII pathway and hematologic disease. Blood. 2012;120(1):31-8.
- Latif A, Aledort L. Inherited combined factor deficiency states. Hemostasis and thrombosis Oxford: Wiley. 2014:127-36.
- Peyvandi F, Menegatti M. Treatment of rare factor deficiencies in 2016. ASH Education Progr
bosis and Haemostasis. 2016;14(2):236-47.
- Oldenburg J, Marinova M, Müller‐Reible C, Watzka M. The vitamin K cycle. Vitamins & Hormones. 2008;78:35-62.
- Oldenburg J, Bevans CG, Müller CR, Watzka M. Vitamin K epoxide reductase complex subunit 1 (VKORC1): the key protein of the vitamin K cycle. Antioxidants & redox signaling. 2006;8(3-4):347-53.
- Gallieni M, Fusaro M. Vitamin K and cardiovascular calcification in CKD: is patient supplementation on the horizon? Kidney international. 2014;86(2):232-4.
- McMillan CW, Roberts HR. Congenital combined deficiency of coagulation factors II, VII, IX and X: report of a case. New England Journal of Medicine. 1966;274(23):1313-5.
- Chung K-S, Bezeaud A, Goldsmith JC, McMillan CW, Menache D, Roberts HR. Congenital deficiency of blood clotting factors II, VII, IX, and X. Blood. 1979;53(4):776-87. am Book. 2016;2016(1):663-9.
- Oukkache B, El Graoui O, Zafad S. Combined factor V and VIII deficiency and pregnancy. International journal of hematology. 2012;96(6):786-8.
- Mansouritorghabeh H, Shirdel A. Desmopressin acetate as a haemostatic elevator in individuals with combined deficiency of factors V and VIII: a clinical trial. Journal of Thrombosis and Haemostasis. 2016;14(2):336-9.
- Brenner B, Kuperman AA, Watzka M, Oldenburg J, editors. Vitamin K–dependent coagulation factors deficiency. Seminars in thrombosis and hemostasis; 2009: © Thieme Medical Publishers.
- Napolitano M, Mariani G, Lapecorella M. Hereditary combined deficiency of the vitamin K-dependent clotting factors. Orphanet journal of rare diseases. 2010;5(1):21.
- Stafford D. The vitamin K cycle. Journal of Thrombosis and Haemostasis. 2005;3(8):1873-8.
- Tie JK, Stafford DW. Structural and functional insights into enzymes of the vitamin K cycle. Journal of Throm
- Brenner B, Sánchez-Vega B, Wu S-M, Lanir N, Stafford DW, Solera J. A missense mutation in γ-glutamyl carboxylase gene causes combined deficiency of all vitamin K-dependent blood coagulation factors. Blood. 1998;92(12):4554-9.
- Oldenburg J, Von Brederlow B, Fregin A, Rost S, Wolz W, Eberl W, et al. Congenital deficiency of vitamin K dependent coagulation factors in two families presents as a genetic defect of the vitamin K-epoxide-reductase-complex. Thrombosis and haemostasis. 2000;84(12):937-41.
- Spronk HM, Farah RA, Buchanan GR, Vermeer C, Soute BA. Novel mutation in the γ-glutamyl carboxylase gene resulting in congenital combined deficiency of all vitamin K–dependent blood coagulation factors. Blood. 2000;96(10):3650-2.
- Lunghi B, Redaelli R, Caimi T, Corno A, Bernardi F, Marchetti G. Novel phenotype and γ‐glutamyl carboxylase mutations in combined deficiency of vitamin K‐dependent coagulation factors. Haemophilia. 2011;17(5):822-4.
- Hauschka PV, Lian JB, Cole D, Gundberg CM. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiological reviews. 1989;69(3):990-1047.
- Pauli R, Lian J, Mosher D, Suttie J. Association of congenital deficiency of multiple vitamin K-dependent coagulation factors and the phenotype of the warfarin e
55-S7.
- Prisco D, Ciuti G, Falciani M. Hemostatic changes in normal pregnancy. Haematologica reports. 2005;1(10):1-5.
- Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. The Lancet. 2013;381(9863):333-42.
- Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best practice & research Clinical endocrinology & metabolism. 2011;25(1):161-mbryopathy: clues to the mechanism of teratogenicity of coumarin derivatives. American journal of human genetics. 1987;41(4):566.
- Vanakker OM, Martin L, Gheduzzi D, Leroy BP, Loeys BL, Guerci VI, et al. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. Journal of Investigative Dermatology. 2007;127(3):581-7.
- Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood. 2015;125(13):2052-61.
- Thomas A, Stirling D. Four factor deficiency. Blood coagulation & fibrinolysis. 2003;14:S79.
- Briggs BJ, Dickerman JD. Bleeding disorders in Noonan syndrome. Pediatric blood & cancer. 2012;58(2):167-72.
- Pfeiffer R, Ott R, Gilgenkrantz S, Alexandre P. Deficiency of coagulation factors VII and X associated with deletion of a chromosome 13 (q34). Evidence from two cases with 46, XY, t (13; Y)(q11; q34). Human genetics. 1982;62(4):358-60.
- Kurosawa H, Suzumura H, Okuya M, Fukushima K, Sugita K, Fujiwara T, et al. Haemostatic management of surgery for imperforate anus in a patient with 13q deletion syndrome with combined deficiency of factors VII and X. Haemophilia. 2009;15(1):398-400.
- Chilcott J, Russell G, Mumford A. Combined deficiency of factors VII and X: clinical description of two cases and management of spinal surgery. Haemophilia. 2006;12(5):555-8.
[1] Thrombin time
[2] Chorionic villus sampling
[3] On-demand therapy
[4] Fresh frozen plasma
[5] Virally-inactivated FFP
[6] 1-desamino-8D-arginine vasopressin
[7] Solvent-detergent FFP
[8] Recombinant FVIII
[9] Vitamin K-dependent coagulation factors deficiency
[10] γ-glutamyl carboxylase
[11] Subunit 1 of vitamin K epoxide reductase complex
[12] Prothrombin complex concentrate
[13] Three trans-membrane model
[14] Post-translational modification
[15] Growth arrest-specific protein 6
[16] Distal digital hypoplasia
[17] Epiphyseal stippling
[18] Warfarin embryopathy
تشخیص آزمایشگاهی و درمان کمبود توأم فاکتورهای انعقادی کمبود مشترک فاکتور V و فاکتور VIII (بخش اول)
روشهای آزمایشگاهی و استانداردهای بینالمللی در تشخیص اختلالات انعقادی (بخش اول)
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام