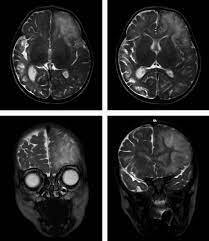
سندرم HHH
Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome

شاهین اسعدی (دانشجوی دکتری تخصصی ژنتیک پزشکی)
کلیات
سندرم هایپرآمونمی، هایپراورنیتینمی، هموسیترولینوری که به اختصار سندرم HHH نیز خوانده می شود و گاهی از آن با نام نقص اورنیتین ترانس لوکاز هم یاد می شود، نوعی اختلال در چرخه اوره است که باعث افزایش سطح آمونیاک در خون می شود. کمبود اُورنیتین ترانس لوکاز، یک اختلال ارثی ژنتیکی است که باعث انباشت آمونیاک در خون می گردد. اگر سطح آمونیاک زمانی که پروتئین ها در بدن تجزیه می شوند، بیش از حد بالا باشد، اثرات سٌمی در پی خواهد داشت. شایان ذکر است که سیستم عصبی به فزونی آمونیاک، بیش از حد حساس است.
علائم و نشانه های بالینی
کمبود اُورنیتین ترانس لوکاز به طور گسترده ای در شدت و سن ابتلاء به آن متفاوت است. نوزادان مبتلا به سندرم HHH دارای سطوح انرژی کم یا فاقد انرژی برای خوردن غذا، کنترل دمای بدن یا کنترل تنفس هستند. برخی از نوزادان مبتلا به این سندرم ممکن است تشنج یا حرکات غیر طبیعی بدن را تجربه کنند که در نهایت، ممکن است به حالت کُما بروند.
در بیشتر افراد مبتلا به سندرم HHH علائم و نشانه های کمبود اُورنیتین ترانس لوکاز تا بعد از زایمان ظاهر نمی شود. ترشح اُورنیتین ترانس لوکاز بعد از متولد شدن شروع می شود که معمولا کمتر از دوره معمول نوزادی است. بعضی از افراد مبتلا به سندرم HHH نمی توانند مواد غذایی حاوی پروتئین بالا مانند گوشت را تحمل (تولرانس) کنند. شایان ذکر است که گاهی اوقات، غذاهایی با پروتئین بالا و یا استرس ناشی از بیماری یا ناشتا بودن ممکن است، سطوح آمونیاک را در خون سریعتر انباشت کنند. این افزایش سریع آمونیاک ممکن است منجر به استفراغ، کاهش انرژی، عدم هماهنگی اندام ها (آتاکسی)، سردرگمی و یا تاری دید شود. علاوه بر این، عوارض ناشی از کمبود اُورنیتین ترانس لوکاز ممکن است شامل تاخیر رشد، معلولیت های یادگیری و اِسپاسم عضلانی باشد.
علت شناسی
سندرم HHH در اثر جهش ژن SLC25A15 که در بازوی بلند کروموزوم شماره 13 به صورت 13q14.11 مستقر است، ایجاد می شود. همانطور که ذکر شد، کمبود اُورنیتین ترانس لوکاز متعلق به یک کلاس از بیماری های ژنتیکی است که به نام اختلالات چرخه اُوره شناخته می شود. چرخه اُوره دنباله ای از واکنش های بیوشیمیایی است که در سلول های کبدی اتفاق می افتد.

شکل 1 : نمای شماتیک از کرومووزم شماره 13 که ژن SLC24A15 در بازوی بلند این کروموزوم به صورت 13q14.11 مستقر است
ژن SLC25A15 دستورالعمل های لازم برای سنتز پروتئینی به نام حمل کننده اُورنیتین میتوکندریایی را فراهم می کند. این پروتئین برای حرکت مولکولی به نام اُورنیتین در میتوکندری (مراکز تولید انرژی در سلول ها) ضروری است. به طور خاص این پروتئین، اُورنیتین را در داخل غشاء درونی میتوکندری به ناحیه ای به نام ماتریکس میتوکندریایی انتقال می دهد که در چرخه اُوره شرکت می کند.
جهش در ژن SLC25A15 منجر به سنتز پروتئین نامناسب یا ناپایدار حمل کننده اُورنیتین میتوکندریایی می شود که نمی تواند اُورنیتین را به ماتریکس میتوکندریایی برساند. این قصور پروتئین حمل کننده اُورنیتین منجر به وقفه چرخه اُوره و انباشت آمونیاک می شود که منجر به علائم و نشانه های سندرم HHH می گردد.
سندرم HHH از الگوی توارثی اتوزومال مغلوب پیروی می کند. بنابراین برای ایجاد این سندرم به دو نسخه از ژن جهش یافته SLC25A15 (یکی از پدر و دیگری از مادر) مورد نیاز است و شانس داشتن فرزندی مبتلا به سندرم HHH در این حالت، برای هر بارداری احتمالی به میزان 25% می باشد.
فراوانی
سندرم HHH یک اختلال ژنتیکی بسیار نادر است که تاکنون کمتر از 100 مورد مبتلا به این سندرم از سراسر جهان در ادبیات پزشکی گزارش شده است.
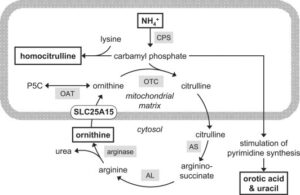
شکل 2 : شماتیکی از مسیر مولکولی ژن SLC25A15
تشخیص
سندرم HHH براساس یافته های بالینی و فیزیکی مبتلایان و برخی آزمایش های پاتولوژیکی، تشخیص داده می شود. آزمایش بررسی سطوح آمونیاک نیز در تشخیص سندرم HHH موثر است. قطعی ترین روش تشخیص سندرم HHH، آزمایش ژنتیک مولکولی برای ژن SLC25A15 به منظور بررسی وجود جهش های احتمالی می باشد.
مسیرهای درمانی
استراتژی درمان و مدیریت سندرم HHH به صورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص تغذیه، متخصص بیوشیمی بالینی، متخصص کبد و گوارش و سایر متخصصان مراقبت های بهداشتی انجام پذیرد. درمان استانداردی برای این سندرم وجود ندارد و تمامی اقدامات بالینی به منظور تخفیف رنج مبتلایان می باشد. مشاوره ژنتیک نیز برای تمامی خانواده هایی که خواستار فرزندی سالم هستند از اهمیت بسزایی برخوردار است.
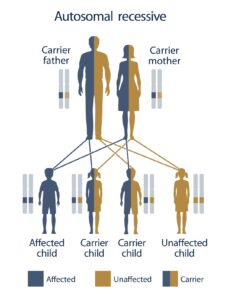
شکل 3 : نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم HHH نیز از این الگو تبعیت می کند
منبع:
اسعدی شاهین و همکاران، کتاب پاتولوژی در ژنتیک پزشکی جلد 5، انتشارات کتب دانشگاهی عمیدی، 1397.
https://medlabnews.ir/%d8%b3%d9%86%d8%af%d8%b1%d9%85-%d8%a7%d8%b3%d9%84%d8%a7%db%8c/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام