سندرم کرونکایت-کانادا
Cronkhite–Canada syndrome

مهسا جمالی (کارشناس ارشد ژنتیک)
سندرم کرونکایت-کانادا (CCS) یک بیماری بسیار نادر است که نشانههای آن شامل از دست دادن طعم و مزه، پولیپ روده، ریزش مو و مشکلات رشد ناخن میباشد. این سندرم در درجه اول، افراد مسن را درگیر میکند (متوسط سن 59 سال) که کمتر از 400 مورد در 50 سال گذشته از این سندرم گزارش شده است و بیشترین فراوانی این سندرم در ژاپن و ایالاتمتحده آمریکا میباشد. سندرم کرونکایت-کانادا با پولیپهای متعدد از دستگاه گوارش همراه است و علت این بیماری قطعیت در ژنتیک و ارثی بودن آن ندارد و بیشتر بهعنوان یک بیماری ایدیوپاتیک (بیماری که علت ناشناخته دارد) شناخته میشود. حدود 70% بیماران مبتلا به این سندرم از تبار ژاپنی هستند و در میان مردم ژاپن، نسبت مرد به زن برای ابتلا به این نوع سندرم، 2 به 1 است.
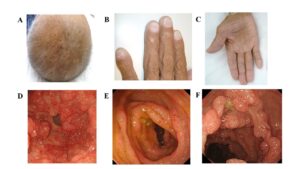
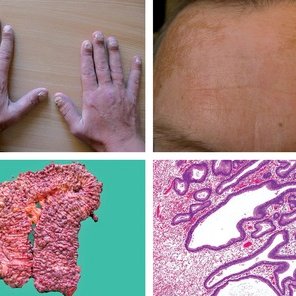
شکل 1: تصاویر ارگانهای مبتلایان سندرم کرونکایت-کانادا
علائم و نشانههای بالینی سندرم کرونکایت-کانادا
نشانههای سندرم کرونکایت-کانادا عبارتاند از: اسهال، گرفتگی عضلات، گاز بیشازحد شکم و درد بیشازحد شکم. این علائم به دلیل پولیپهای متعدد در جداره معده، روده کوچک و روده بزرگ است. افراد مبتلا به سندرم کرونکایت-کانادا ممکن است قادر به هضم شیر و فراوردههای لبنی (عدم تحمل لاکتوز) نباشند و همچنین با کاهش توانایی لازم در جذب مواد مغذی (سوء جذب) از طریق روده کوچک بهصورت صحیح مواجه باشند. حدود 15% از افراد مبتلا به سندرم کرونکایت-کانادا در نهایت به توسعه سرطان روده بزرگ دچار میشوند. افراد مبتلا به سندرم کرونکایت-کانادا ممکن است بهطور غیرطبیعی سطوح پائینی از پروتئین در خون را داشته باشند. در افراد مبتلا به سندرم کرونکایت-کانادا ممکن است به دلیل اسهال مزمن، عدم تعادل الکترولیتها وجود داشته باشد. برخی دیگر از مبتلایان سندرم کرونکایت کانادا، ممکن است یک کبودی بزرگ روی پوست به فرم یک برچسب را داشته باشند و یا ممکن است عملکرد طبیعی ریه مختل شود. دیگر نشانههای این سندرم شامل: از دست دادن مو (آلوپسی)، پدیدار شدن لکههای تیره در مناطق زیادی روی پوست (هایپرپیگمانتاسیون)، تغییرات دژنراتیو و از دست دادن ناخن میباشد.
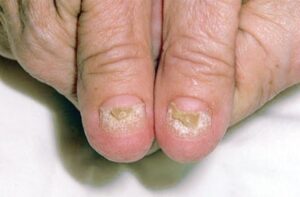
شکل 2: تصویر فرد مبتلا به سندرم کرونکایت-کانادا، به ناخنهای پوسیده انگشتان دست دقت کنید.
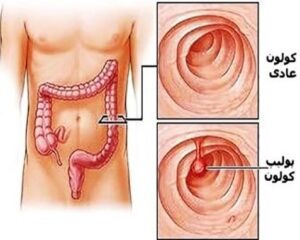
شکل 3: نمای شماتیک از روده نرمال در مقایسه با روده دارای پولیپ
فرکانس سندرم کرونکایت-کانادا
سندرم کرونکایت-کانادا اختلال بسیار نادری است که زنان را بیشتر از مردان تحت تأثیر قرار میدهد. نسبت زنان مبتلا به سندرم کرونکایت-کانادا، حدود 1.5 برابر مردان است. بهطورمعمول، سن شروع این بیماری در میانسالی است که متوسط آن حدود 59 سال است و میتواند با طیف وسیعی از انواع علائم مربوطه از 31 سالگی تا 85 سالگی آغاز گردد.
تشخیص سندرم کرونکایت-کانادا
تشخیص این سندرم بر اساس یافتههای بالینی و کلینیکی از بیماران که شامل: اسهال مکرر و بیدلیل، درد بیشازحد شکم در ساعات مختلف شبانهروز، وجود پولیپهای متعدد در جداره روده بدون سابقه ژنتیکی، گرفتگی بیسابقه عضلات، ریزش موی سر و بدن بدون سابقه خانوادگی، وجود لکههای تیره به رنگ قهوهای در مناطق عمده پوست و کاهش مقاومت ناخنها تا جایی که خودبهخود تخریب شوند، انجام میگیرد. برخی از علائم این سندرم با دیگر آسیبهای ژنتیکی انسان مانند سندرم پوتز-جگرز، سندرم تورکات، سندرم گاردنر مشابه است که بر اساس نوع پولیپ ایجاد شده در جداره روده، نوع سندرم نیز افتراق پیدا میکند؛ بنابراین نبایستی صرفاً با مشاهده پولیپ در جداره روده، سندرم کرونکایت-کانادا را به قطعیت اعلام کرد.
مسیرهای درمانی سندرم کرونکایت-کانادا
ازآنجاکه علت این سندرم، خیلی کم شناسایی شده است، درمان نیز بر اساس علامت است. به عبارتی بر پایه نشانهها و علامتی که بیماران مبتلا به سندرم کرونکایت-کانادا از خود نشان میدهند، نوع مسیر درمان یا کنترل اختلال مربوطه در حد امکان، متفاوت خواهد بود. یکی از این مسیرها، استفاده از مکملهای غذایی و رژیم تغذیهای مناسب بهمنظور جبران پروتئینهای ازدسترفته، متعادل کردن جذب روده از مواد مغذی و متعادل کردن مایعات و الکترولیتهای بدن میباشد. داروهای استروئیدی مثل پردنیزون، نیز گاهی اوقات برای کاهش التهاب روده در مبتلایان سندرم کرونکایت-کانادا استفاده میشود. رشد بیشازحد باکتریها در روده که باعث سوء جذب میشوند را میتوان با آنتیبیوتیک درمان کرد. در موارد نادر، بدون هیچ دلیل روشن و مستندی، علائم سندرم کرونکایت-کانادا خودبهخود بهبود مییابد. عمل جراحی نیز برای برداشت پولیپ، میتواند به کاهش برخی علائم سندرم کرونکایت-کانادا کمک کند.
:References
- Vernia P, Marcheggiano A, Marinaro V, Morabito S, Guzzo I, Pierucci A (October 2005).
- Calva D, Howe JR (August 2008). “Hamartomatous polyposis syndromes”. The Surgical clinics of North America. 88 (4): 779–817, vii.
- Cronkhite LW, Canada WJ (June 1955). “Generalized gastrointestinal polyposis; an unusual syndrome of polyposis, pigmentation, alopecia and onychotrophia”. N. Engl. J. Med. 252 (24): 1011–5.
- Junnarkar SP, Sloan JM, Johnston BT, Laird JD, Irwin ST (May 2001). “Cronkhite-Canada syndrome”. The Ulster medical journal. 70 (1): 56–8.
- Nagata J, Kijima H, Hasumi K, Suzuki T, Shirai T, Mine T (June 2003). “Adenocarcinoma and multiple adenomas of the large intestine, associated with Cronkhite-Canada syndrome”. Dig Liver Dis. 35 (6): 434–8.
- Ward E, Wolfsen HC, Ng C (February 2002). “Medical management of Cronkhite-Canada syndrome”. South. Med. J. 95 (2): 272–4.
- Oberhuber G, Cronkhite-Canada Syndrome. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:338.
- Yamada T, Alpers DH, Owyang C, et al. Eds. Textbook of Gastroenterology. 2nd ed. J. B. Lippincott Company. Philadephia, PA; 1995:1960.
- Ward EM, Wolfsen HC. Review Article: the non-inherited gastrointestinal polyposis syndromes. Aliment Pharmacol Ther. 2002;16:333-42.
- Egawa T, Kubota T, Otani Y, et al. Surgically treated Cronkhite-Canada syndrome associated with gastric cancer. Gastric Cancer. 2000;27:156-60.
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام