سندرم لنفوپرولیفراتیو اُتوایمیون
Autoimmune Lymphoproliferative Syndrome
(ALP Syndrome)

شاهین اسعدی (دانشجوی دکتری تخصصی ژنتیک پزشکی)
کلیاتی از سندرم لنفوپرولیفراتیو اُتوایمیون (ALP)
سندرم لنفوپرولیفراتیو اُتوایمیون یک اختلال ژنتیکی ارثی است که در آن، بدن قادر به تنظیم صحیح سلولهای سیستم ایمنی (لنفوسیتها) نیست. این سندرم با تولید تعداد زیادی از لنفوسیتهای غیرطبیعی (لنفوپرولیفراسیون) مشخص میشود. تجمع لنفوسیتهای اضافی باعث افزایش غدههای لنفاوی (لنفادنوپاتی)، بزرگی کبد (هپاتومگالی) و بزرگی طحال (اسپیلنومگالی) میشود.
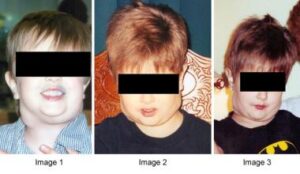
شکل 1: تصاویر کودکان مبتلا به سندرم لنفوپرولیفراتیو اُتوایمیون (ALP) همراه با اختلال مربوطه در ناحیه گردن
علائم و نشانههای بالینی سندرم لنفوپرولیفراتیو اُتوایمیون (ALP)
در افراد مبتلا به سندرم لنفوپرولیفراتیو اُتوایمیون خطر ابتلا به سرطان سلولهای سیستم ایمنی بدن (لنفوم) افزایش مییابد و همچنین ممکن است خطر ابتلا به سرطانهای دیگر نیز افزایش یابد. علاوه بر این، اختلالات خودایمنی نیز در سندرم ALP رایج هستند. اختلالات خودایمنی زمانی رخ میدهند که سیستم ایمنی بدن به بافتها و اندامهای خودی حمله کند و آنها را تخریب سازد. هدف بیشتر اختلالات خودایمنی مرتبط با سندرم ALP، آسیب رساندن به سلولهای خونی است؛ به عنوان مثال سیستم ایمنی بدن ممکن است به گلبولهای قرمز خون (آنمی همولیتیک اُتوایمیون)، گلبول سفید (نوتروپنی اُتوایمیون) یا پلاکتها (ترومبوسایتوپنی اُتوایمیون) حمله کند. بهطور معمول، اختلالات خودایمنی که بر اندام و بافتهای دیگر تأثیر میگذارد، میتواند در افراد مبتلا به سندرم ALP رخ دهد. این اختلالات میتوانند به کلیهها (گلومرولونفریت)، کبد (هپاتیت اُتوایمیون)، چشمها (یوئیت)، اعصاب (سندرم گلین- بار) یا بافت همبند (لوپوس اِریتماتوز سیستمیک) آسیب برسانند.
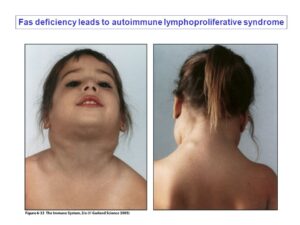
شکل 2: تصاویر دیگر از کودک مبتلا به سندرم لنفوپرولیفراتیو اُتوایمیون (ALP) همراه با اختلالات مربوطه
مشکلات پوستی مانند راش یا کهیر نیز میتواند در سندرم ALP رخ دهد. گاهی اوقات افراد مبتلا به سندرم ALP دارای ضایعات پوستی همراه با درد (پانیکولیت) هستند. سایر علائم و نشانههای نادر در سندرم ALP عبارتند از: التهاب مفصلی (آرتریت)، التهاب عروق خونی (واسکولیت)، زخم دهان و یا نارسایی زودرس تخمدان. علاوه بر این، افراد آسیبدیده با سندرم ALP ممکن است آسیب عصبی را با علائمی که شامل سردرد، تشنج و یا زوال عقلی هستند، نیز تجربه کنند.
علتشناسی سندرم لنفوپرولیفراتیو اُتوایمیون (ALP)
تقریباً 75 درصد از موارد سندرم لنفوپرولیفراتیو اُتوایمیون در اثر جهش ژن FAS که در بازوی بلند کروموزوم شماره 10 بهصورت 10q23.31 مستقر است، ایجاد میشود. این ژن دستورالعملهای لازم برای سنتز پروتئین درگیر در سیگنالهای سلولی را فراهم میکند که منجر به خودکشی سلولها (آپوپتوز) میشود.

شکل 3: نمای شماتیک از کروموزوم شماره 10 که ژن FAS در بازوی بلند این کروموزوم بهصورت 10q23.31 مستقر است
زمانی که سیستم ایمنی، بدن را برای مبارزه با عفونت فعال میکند، تعداد زیادی از لنفوسیتها تولید میشود. در حالت عادی، هنگامی که این لنفوسیتها دیگر نیاز نباشند، دچار آپوپتوزیس میشوند. جهش در ژن FAS منجر به تولید پروتئین غیرطبیعی میشود که با فرآیند آپوپتوز تداخل ایجاد میکند؛ بنابراین لنفوسیتهای اضافی که دچار آپوپتوز نشدهاند در بافتها و اندامهای بدن تجمع مییابند و اغلب به آنها تهاجم میکنند و منجر به ایجاد اختلالات خودایمنی میشوند. تداخل با فرآیند آپوپتوزیس به سلولها اجازه میدهد تا بدون کنترل تکثیر شوند و منجر به لنفوم و سرطانهای دیگر در افراد مبتلا به سندرم ALP شوند. شایان ذکر است که سندرم ALP ممکن است در اثر جهش ژنهای دیگری نیز ایجاد شود که هنوز شناسایی نشدهاند.
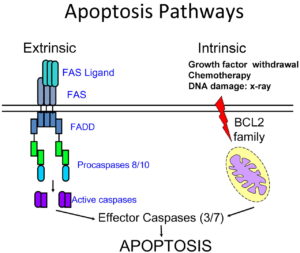
شکل 4: شماتیکی از مسیر مولکولی ژن FAS در آبشار القایی مرگ فیزیولوژیک سلولی (آپوپتوز)
سندرم ALP در اکثر موارد با جهش در ژن FAS از الگوی توارثی اتوزومال غالب پیروی میکند؛ بنابراین برای ایجاد این سندرم، یک نسخه از ژن جهشیافته FAS (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزند مبتلا به سندرم ALP در حالت اتوزومی غالب، برای هر بارداری احتمالی به میزان 50% است. در موارد نادر، سندرم ALP با جهش در ژن FAS از الگوی توارثی اتوزومال مغلوب پیروی میکند؛ بنابراین برای ایجاد این سندرم، دو نسخه از ژن جهشیافته FAS (یکی از پدر و دیگری از مادر) موردنیاز است و شانس داشتن فرزند مبتلا به سندرم ALP در حالت اتوزومی مغلوب، برای هر بارداری احتمالی به میزان 25% است.
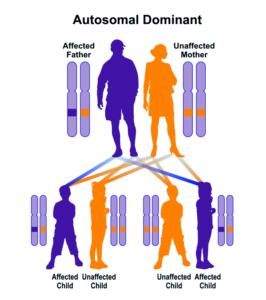
شکل 5: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم ALP در اکثر موارد از این الگو تبعیت میکند
فراوانی سندرم لنفوپرولیفراتیو اُتوایمیون (ALP)
سندرم ALP اختلال ایمونوژنتیکی نادر است که فرکانس شیوع آن در جهان مشخص نیست. تاکنون بیش از 200 مورد مبتلا به این سندرم از سراسر جهان در ادبیات پزشکی گزارش شده است.
تشخیص سندرم لنفوپرولیفراتیو اُتوایمیون (ALP)
سندرم ALP بر اساس یافتههای بالینی مبتلایان و برخی آزمایشهای پاتولوژیکی، تشخیص داده میشود. دقیقترین روش تشخیص این سندرم، آزمایش ژنتیک مولکولی برای ژن FAS به منظور بررسی وجود جهشهای احتمالی است.
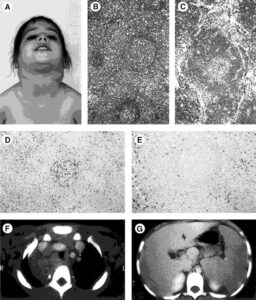
شکل 6: تصاویر رادیولوژیکی و میکروسکوپی از اختلالات مربوط به سندرم ALP در کودک مبتلا
مسیرهای درمانی سندرم لنفوپرولیفراتیو اُتوایمیون (ALP)
استراتژی درمان و مدیریت سندرم ALP بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص هماتولوژی، متخصص اُنکولوژی، ایمونولوژیست و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان استانداردی برای سندرم ALP وجود ندارد و تمامی اقدامات بالینی به منظور کاهش رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند، از اهمیت بسزایی برخوردار است.
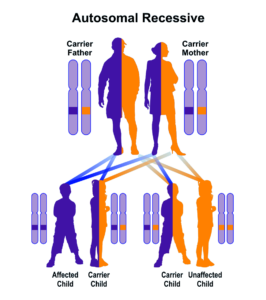
شکل 7: نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم ALP نیز در برخی موارد از این الگو میتواند تبعیت کند
منبع:
اسعدی شاهین و همکاران، کتاب پاتولوژی در ژنتیک پزشکی جلد 5، انتشارات کتب دانشگاهی عمیدی، 1397.
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام