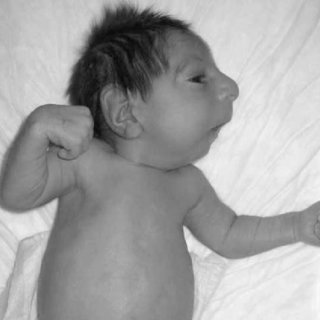
سندرم سکل
Seckel Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، محمدحسین مبلغ حسینی (کارشناس علوم آزمایشگاهی، دانشجوی کارشناسی ارشد ژنتیک)، ویدا وحدانی (کارشناس مامایی، دانشجوی کارشناسی ارشد ژنتیک)

نگارنده مسئول: شاهین اسعدی (Molecular Geneticist)
کلیات
سندرم سکل یا کوتولگی میکروسفالیک، یک اختلال ژنتیکی بسیار نادر است که با تأخیر رشد قبل از تولد و وزن کم هنگام تولد، همراه است. تأخیر رشد پس از تولد نیز ادامه پیدا میکند که منجر به کوتاهی قد و کوتولگی میشود. دیگر نشانهها و ویژگیهای فیزیکی مرتبط با سندرم سکل شامل: اندازه سر غیرعادی کوچک (میکروسفالی)، درجه مختلفی از عقبماندگی ذهنی، بینی برآمده و نوک بینی ظاهری منقار مانند، چشم غیرطبیعی بزرگ، چهره باریک، گوش ناقص، فک کوچک غیرمعمول، خمیدگی انگشتان پنجم، دیسپلازی باسن، دررفتگی استخوان ساعد و دیگر اختلالات فیزیکی میباشد.

شکل 1: تصویر کودک مبتلا
علائم و نشانهها
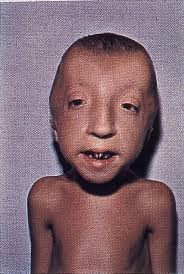
سندرم سکل با رشد غیرطبیعی و آهسته جنین (عقبماندگی رشد داخل رحمی) مشخص میشود که در نتیجه وزن نوزادان مبتلا به سندرم سکل، هنگام تولد کمتر از حد نرمال میشود. پس از تولد نیز، رشد استخوانی مبتلایان سندرم سکل به تأخیر میافتد که منجر به کوتاهی قد و کوتولگی میشود. عقبماندگی ذهنی متوسط تا شدید نیز ممکن است در نوزادان مبتلا به این سندرم هنگام تولد رخ دهد، اما ممکن است در ظاهر مشخص نشود و تا زمانی که نوزاد مبتلا مسنتر شود، عقبماندگی ذهنی نیز آشکار خواهد شد.

شکل 2: تصویر دختر مبتلا همراه با کوتولگی مادرزادی.
علاوه بر این، نوزادان مبتلا اختلالات متمایز از سروصورت را تجربه خواهند کرد. در اغلب موارد، نوزادان مبتلا به سندرم ممکن است علائمی مانند اندازه دور سر کوچکتر از حد نرمال و سن فیزیولوژیک مبتلایان (میکروسفالی)، پیشانی کوچک، فک کوچک غیرمعمول (میکروگناتیا) و نوک بینی به شکل منقار را آشکار کنند. علاوه بر این، در برخی موارد از سندرم سکل ممکن است استخوانهای لیفی جمجمه (شکاف جمجمه) زودتر از موعد مقرر جوش بخورند که منجر به دراز یا کوتاه شدن شکل سر، بسته به قسمتی از جمجمه که تحت تأثیر قرار میگیرد، میشود.

شکل 3: نمایی دیگر از دختر مبتلا به سندرم با کوتولگی مادرزادی.
در برخی از نوزادان مبتلا به سندرم سکل، سایر اختلالات سروصورت ممکن است وجود داشته باشد از جمله چشم غیرطبیعی بزرگ، شکاف پلکی، لوچی چشم (استرابیسم)، گوشهای ناقص (دیسپلاستیک) با عدم وجود نرمه گوش، سقف بسیار قوس از دهان (شکاف کام)، یکطرف صورت بزرگتر از طرف دیگر (عدم تقارن صورت)، هایپوپلازی مینای دندان، ازدحام و یا موقعیت نامناسب دندانها در دهان.
علاوه بر این، برخی از کودکان مبتلا به سندرم سکل ممکن است اختلالات اسکلتی مانند دررفتگی استخوان ساعد، دررفتگی آرنج، دررفتگی باسن و یا ناتوانی بهطور کامل از تحرک زانو را تجربه کنند. همچنین ممکن است انحنای غیرطبیعی ستون فقرات (کیفواسکولیوز)، خمیدگی انگشتان پنجم، پاچنبری و یا عدم وجود یکی از دندهها در سندرم سکل رخ دهد. در برخی موارد، مردان مبتلا به سندرم سکل ممکن است، عدم فرود بیضهها به داخل کیسه بیضه را تجربه کنند و زنان مبتلا به این سندرم نیز ممکن است چوچوله غیرطبیعی بزرگ شده (کلیتورومگالی) را تجربه کنند. علاوه بر این، کودکان مبتلا به سندرم سکل ممکن است رشد بیشازحد مو در بدن (هایرسوتیسم) و یا چینخوردگی عمیق در کف دست را آشکار کنند.
در برخی از موارد این سندرم، اختلالات خون (هماتولوژی) از جمله کمبود عناصر مغز استخوان مثل سلولهای قرمز خون (اریتروسیت) و سلولهای سفید خون (لکوسیت) و پلاکتها نیز گزارش شده است.

شکل 4: تصویر کودک مبتلا همراه با لاغری بیشازحد و کوتولگی مادرزادی.
علت شناسی
محققان سندرم سکل را به چهار نوع تقسیمبندی کردهاند که عبارتاند از: نوع 1 (SCKL1)، نوع 2 (SCKL2)، نوع 3 (SCKL3) و نوع 4 (SCKL4).
سندرم نوع 1 (SCKL1) در اثر جهش ژن ATR که در بازوی بلند کروموزوم شماره 3 بهصورت 3q23 مستقر است، ایجاد میشود. سندرم نوع 2 (SCKL2) در اثر جهش ژن RBBP8 که در بازوی بلند کروموزوم شماره 18 بهصورت 18q11.2 مستقر است، ایجاد میشود. سندرم نوع 3 (SCKL3) در اثر جهش ژن SCKL3 که در بازوی بلند کروموزوم شماره 14 بهصورت 14q23 مستقر است، ایجاد میشود. سندرم نوع 4 (SCKL4) در اثر جهش ژن CENPJ که در بازوی بلند کروموزوم شماره 13 بهصورت 13q12.12-q12.13 مستقر است، ایجاد میشود. سندرم سکل در تمامی حالتها از الگوی توارثی اتوزومال مغلوب پیروی میکند، بنابراین برای ایجاد این سندرم به دو نسخه از ژنهای جهشیافته ATR، RBBP8،SCKL3 ، CENPJ(یکی از پدر و دیگری از مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به سندرم سکل برای هر بارداری احتمالی، به میزان 25% میباشد.
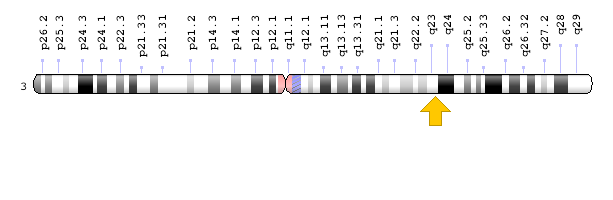
شکل 5: نمای شماتیک از کروموزوم شماره 3 که ژن ATR در بازوی بلند این کروموزوم بهصورت 3q23 مستقر است.
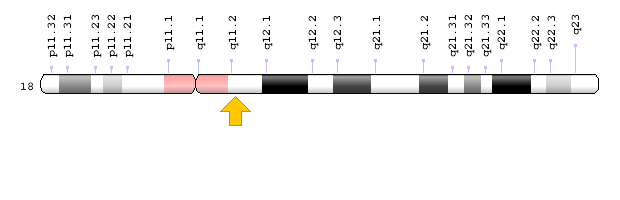
شکل 6: نمای شماتیک از کروموزوم شماره 18 که ژن RBBP8 در بازوی بلند این کروموزوم بهصورت 18q11.2 مستقر است.
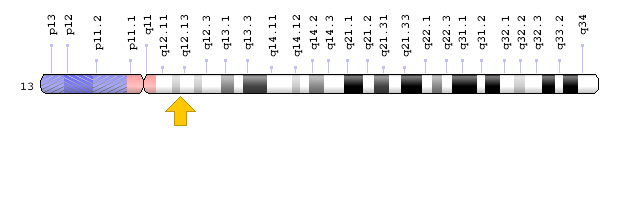
شکل 7: نمای شماتیک از کروموزوم شماره 14 که ژن SCKL3 در بازوی بلند این کروموزوم بهصورت 14q23 مستقر است.
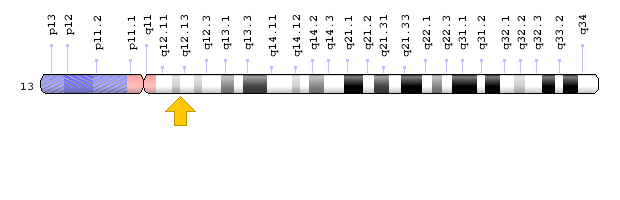
شکل 8: نمای شماتیک از کروموزوم شماره 13 که ژن CENPJ در بازوی بلند این کروموزوم بهصورت 13q12.12-q12.13 مستقر است.
فراوانی
سندرم سکل اختلال ژنتیکی بسیار نادری است که به نظر میرسد مردان و زنان را به تعداد مساوی تحت تأثیر قرار میدهد. از سال 1960 تاکنون بیش از 100 مورد از سندرم سکل در ادبیات پزشکی گزارش شده است. تعیین فرکانس واقعی این سندرم به دلیل عدمتشخیص صحیح، در جمعیت عمومی جهان دشوار است.
تشخیص
تشخیص این سکل بر اساس یافتههای فیزیکی مشخص و ارزیابی دقیق علائم بالینی مبتلایان و شرححال سابقه خانوادگی بیمار، انجام میپذیرد. بهکارگیری تصویربرداری رادیولوژی مانند ام.آر.آی و اشعه ایکس در تشخیص اختلالات اسکلتی سندرم سکل میتواند کمککننده باشد. تشخیص قبل از تولد نیز با استفاده از سونوگرافی جنین مبنی بر ارزیابی میزان رشد جنین امکانپذیر است. بااینحال، قطعیترین روش تشخیص سندرم سکل آزمایش ژنتیک مولکولی برای ژنهای ATR,RBBP8,SCKL3,CENPJ بهمنظور ارزیابی وجود جهشهای احتمالی میباشد.
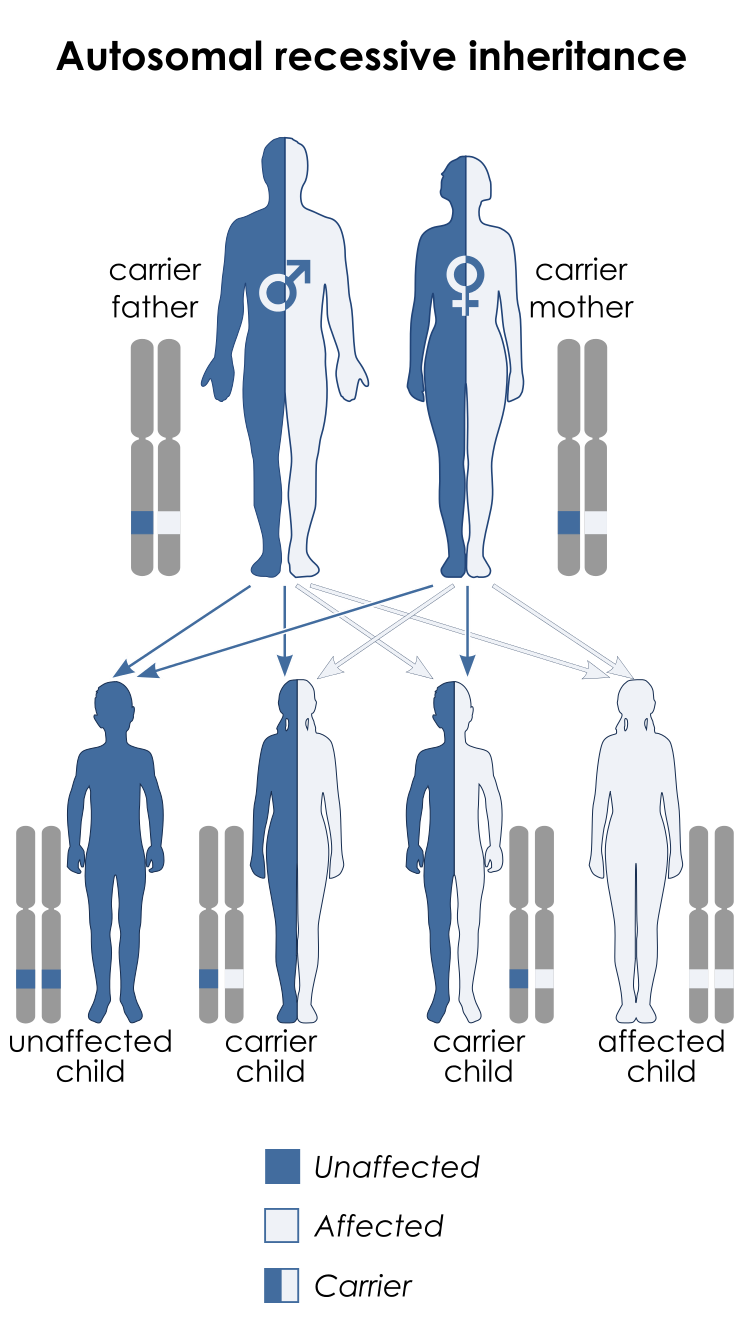
شکل 9: نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم سکل نیز از این الگو تبعیت میکند.
مسیرهای درمانی
سندرم سکل توسط علائم و نشانههایی که هر فرد مبتلا آن را آشکار میشود، اقدام به درمان میشود. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص ارتوپدی، متخصص هورمون و رشد، متخصص تغذیه و متخصص مغز و اعصاب انجام گیرد. متأسفانه هیچ درمان قاطعی برای سندرم سکل وجود ندارد و مبتلایان این اختلال اکثراً از کوتولگی خود رنج میبرند. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم و نرمال هستند از اهمیت ویژهای برخوردار است.
تاریخچه
سندرم سکل اولین بار در سال 1960 توسط دکتر Helmut Paul Georg Seckel پزشک آمریکایی گزارش گردید.

شکل 10: تصویر دکتر Helmut Paul Georg Seckel کاشف سندرم سکل در سال 1960.
منابع:
اسعدی شاهین، جمالی مهسا، باقری رعنا، سادهدل سمانه، توحیدی راد مانوش، کتاب پاتولوژی در ژنتیک پزشکی جلد دوم (M-Z), صفحات 1105-1099, انتشارات کتب دانشگاهی عمیدی، بهار 1396.
سندرم کمخونی دیاموند- بلکفان Diamond-Blackfan Anemia Syndrome
سندرم زلوگر Zellweger Syndrome
https://rarediseases.info.nih.gov/diseases/8562/seckel-syndrome
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام