
سندرم ایکاردی-گاتیئرز
Aicardi–Goutières Syndrome

شاهین اسعدی (دانشجوی ژنتیک مولکولی)، مهسا جمالی (کارشناس ارشد ژنتیک)، مانوش توحیدی راد (کارشناس ارشد ژنتیک)، رعنا باقری (کارشناس ارشد ژنتیک)
مؤلف مسئول: شاهین اسعدی (Molecular Geneticist)
کلیاتی از سندرم ایکاردی-گاتیئرز
سندرم ایکاردی-گاتیئرز، یک اختلال ژنتیکی نادر است که معمولاً در اوایل دوران کودکی آغاز میشود و با التهاب مغز و پوست (اختلال عصبی پوستی) همراه است. اکثر افراد مبتلا به سندرم ایکاردی-گاتیئرز، مشکلات فکری و جسمی قابلتوجهی را تجربه میکنند، اگرچه همیشه چنین نیست. ویژگیهای بالینی در زنان مبتلا به سندرم ایکاردی-گاتیئرز، مشابه عفونتهای رحمی بوده و با ویژگیهای بیماری خودایمنی لوپوس اریتماتوی سیستمیک همپوشانی دارد.
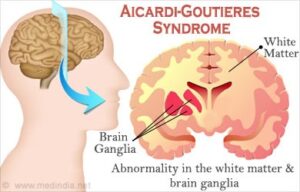
شکل 1: نمای شماتیک از مسیر اثرگذاری سندرم ایکاردی-گاتیئرز در سلولهای مغز انسان
علائم و نشانههای سندرم ایکاردی-گاتیئرز
همانطور که ذکر شد، سندرم ایکاردی-گاتیئرز یک اختلال ژنتیکی نادر است که بهطور عمده بر مغز، سیستم ایمنی بدن و پوست تأثیر میگذارد. بیشتر نوزادان مبتلا به سندرم ایکاردی-گاتیئرز هیچ علائم و نشانهای را از این اختلال در بدو تولد، آشکار نمیکنند، با این حال، در حدود 20 درصد از نوزادان مبتلا به این سندرم با ترکیبی از ویژگیهایی که شامل بزرگ شدن کبد و طحال (بزرگی کبد و طحال)، سطح بالای آنزیمهای کبدی در خون، کاهش پلاکت خون (ترومبوسیتوپنی) و پاسخهای عصبی غیرطبیعی میباشد، متولد میشوند. این ترکیب از علائم و نشانهها معمولاً با پاسخ سیستم ایمنی بدن به عفونت مادرزادی ویروسی همراه است، اما در نوزادان مبتلا به سندرم ایکاردی-گاتیئرز بدون عفونتهای ویروسی، این علائم آشکار میشود.
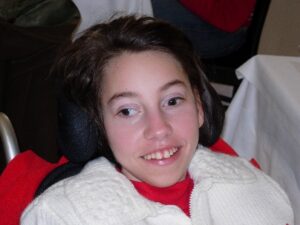
شکل 2: تصویر دختر مبتلا به سندرم ایکاردی-گاتیئرز همراه با اختلالات چشم (استرابیسم) و دندان
بیشتر افراد مبتلا در سال اول زندگی، اختلال شدیدی را در یک قسمت مغز (انسفالوپاتی) که معمولاً برای چند ماه پایدار است، تجربه میکنند. در طول این مرحله، نوزادان معمولاً بسیار تحریکپذیر بوده و خوب تغذیه نمیکنند. همچنین این نوزادان ممکن است بهطور متناوب در غیاب عفونت دچار تب شوند و تشنج بگیرند، علاوه بر این در نوزادان مبتلا رشد مغز و جمجمه کند میشود و در نتیجه اندازه سر بهطور غیرعادی کوچک (میکروسفالی) خواهد بود.
در موارد شدید سندرم ای کاردی-گاتیئرز، ماده سفید مغز که بهعنوان عایق و محافظ اعصاب است، بهطور کامل توسعه پیدا نمیکند و یا وجود ندارد (لکودیستروفی) و افراد مبتلا به این سندرم، تجمع غیرطبیعی کلسیم (کلسیفیکاسیون) را در مغز تجربه خواهند کرد. اغلب افراد مبتلا به سندرم ایکاردی-گاتیئرز، ناتوانی عمیق ذهنی دارند و همچنین مشکلات عصبی و عضلانی قابلتوجهی از جمله سفتی عضلات (اسپاسم) را آشکار میکنند.

شکل 3: تصویر مبتلایان سندرم ایکاردی-گاتیئرز (چهار نفر نشسته) همراه با اختلالات مشترک
بهعلاوه، حدود 40 درصد از مبتلایان سندرم ایکاردی-گاتیئرز، ضایعات پوستی خارشدار دردناک در انگشتان دست و پا و گوش را تجربه میکنند. این ضایعات به شکل پفکرده بوده و قرمز رنگ میباشند که توسط التهاب رگهای خونی کوچک ایجاد میشوند، همچنین مشکلات بینایی از جمله استرابیسم یا لوچی چشم، سفتی مفاصل، عدم توسعه ناخن در پاها و زخمهاای دهان نیز ممکن است در سنــــــدرم ایکاردی-گاتیئرز رخ دهد.
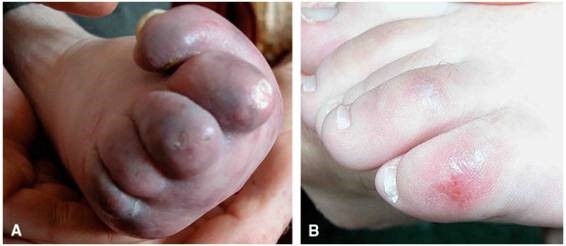
شکل 4: تصاویر پای کودک مبتلا به سندرم ایکاردی-گاتیئرز همراه با ضایعات خارشدار قرمز
شایان ذکر است که بسیاری از افراد مبتلا به سندرم ایکاردی-گاتیئرز، در دوران کودکی جان خود را از دست میدهند، اما ممکن است برخی از مبتلایان با علائم خفیف تا بزرگسالی زنده بمانند.
علتشناسی سندرم ایکاردی-گاتیئرز
سندرم ایکاردی-گاتیئرز به هفت نوع تقسیمبندی میشود که در اثر جهش ژنهای TREX1، RNASEH2A،RNASEH2B ،RNASEH2C ،SAMHD1 ، ADAR1 و IFIH1 ایجاد میشود.
ژن TREX1 در بازوی کوتاه کروموزوم شماره 3 بهصورت 3p21.31 مستقر است.
ژن RNASEH2A در بازوی کوتاه کروموزوم شماره 19 بهصورت 19p13.13 مستقر است.
ژن RNASEH2B در بازوی بلند کروموزوم شماره 13 بهصورت 13q14.3 مستقر است.
ژن RNASEH2C در بازوی بلند کروموزوم شماره 11 بهصورت 11q13.1 مستقر است.
ژن SAMHD1 در بازوی بلند کروموزوم شماره 20 بهصورت 20q11.23 مستقر است.
ژن ADAR1 در بازوی بلند کروموزوم شماره 1 بهصورت 1q21.3 مستقر است.
ژن IFIH1 در بازوی بلند کروموزوم شماره 2 بهصورت 2q24.2 مستقر است.
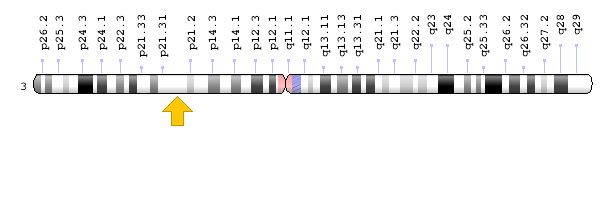
شکل 5: نمای شماتیک از کروموزوم شماره 3 که ژن TREX1 در بازوی کوتاه این کروموزوم بهصورت 3p21.31 مستقر است
ژنهای TREX1، RNASEH2A،RNASEH2B و RNASEH2C حاوی دستورالعمل لازم برای سنتز آنزیم نوکلئاز هستند که در جلوگیری از خطاهای احتمالی مولکول DNA و RNA نقش مهمی را ایفا میکند. ژنهای SAMHD1، ADAR1 و IFIH1 حاوی دستورالعمل لازم برای سنتز پروتئینهای سیستم ایمنی در پاسخ به پروسههای التهابی میباشند، بنابراین جهش در این ژنها منجر به اختلال سیستم ایمنی در پاسخ به عفونتها یا التهابها و اختلال در خطاهای احتمالی مولکول DNA و RNA خواهد شد.
 شکل 6: نمای شماتیک از کروموزوم شماره 19 که ژن RNASEH2A در بازوی کوتاه این کروموزوم بهصورت 19p13.13 مستقر است
شکل 6: نمای شماتیک از کروموزوم شماره 19 که ژن RNASEH2A در بازوی کوتاه این کروموزوم بهصورت 19p13.13 مستقر است
شکل 7: نمای شماتیک از کروموزوم شماره 13 که ژن RNASEH2B در بازوی بلند این کروموزوم بهصورت 13q14.3 مستقر است
شکل 8: نمای شماتیک از کروموزوم شماره 11 که ژن RNASEH2C در بازوی بلند این کروموزوم بهصورت 11q13.1 مستقر است
شکل 9: نمای شماتیک از کروموزوم شماره 20 که ژن SAMHD1 در بازوی بلند این کروموزوم بهصورت 20q11.23 مستقر است
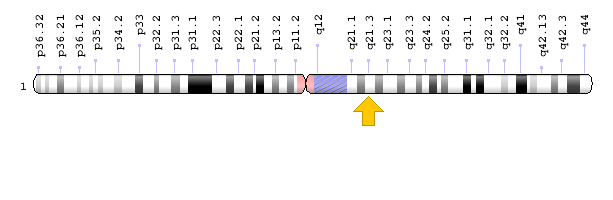
شکل 10: نمای شماتیک از کروموزوم شماره 1 که ژن ADAR1 در بازوی بلند این کروموزوم بهصورت 1q21.3 مستقر است
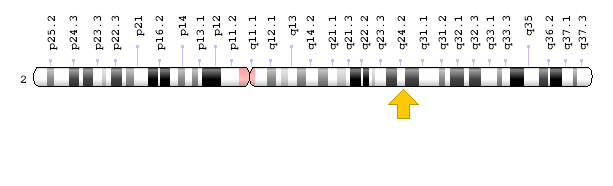 شکل 11: نمای شماتیک از کروموزوم شماره 2 که ژن IFIH1 در بازوی بلند این کروموزوم بهصورت 2q24.2 مستقر است
شکل 11: نمای شماتیک از کروموزوم شماره 2 که ژن IFIH1 در بازوی بلند این کروموزوم بهصورت 2q24.2 مستقر است
سندرم ایکاردی-گاتیئرز از الگوهای وراثتی مختلفی میتواند پیروی کند، با این حال در اغلب موارد از الگوی توارثی اتوزومال مغلوب پیروی میکند، بنابراین برای ایجاد این سندرم با الگوی وراثتی مغلوب، دو نسخه از ژنهای جهشیافته مذکور (یکی از پدر و دیگری از مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به سندرم ایکاردی-گاتیئرز در این حالت، برای هر بارداری احتمالی به میزان 25% میباشد.
در موارد نادر، سندرم ایکاردی-گاتیئرز از الگوی توارثی اتوزومال غالب پیروی میکند، بنابراین برای ایجاد این سندرم با الگوی وراثتی غالب، تنها یک نسخه از ژنهای جهشیافته ذکرشده (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به سندرم ایکاردی-گاتیئرز در این حالت، برای هر بارداری احتمالی به میزان 50% میباشد.
جدول شماره 1: انواع سندرم ایکاردی-گاتیئرز همراه با ژنهای مسئول و محل استقرار آنها و میزان فراوانی در بروز این اختلال
| نوع سندرم | ژن مسئول | جایگاه ژن | فراوانی سندرم |
| ایکاردی-گاتیئرز 1 | TREX1 | 3p21.31 | 23% (1% غالب) |
| ایکاردی-گاتیئرز 2 | RNASEH2B | 13q14.3 | 36% |
| ایکاردی-گاتیئرز 3 | RNASEH2C | 11q13.1 | 12% |
| ایکاردی-گاتیئرز 4 | RNASEH2A | 19p13.13 | 5% |
| ایکاردی-گاتیئرز 5 | SAMHD1 | 20q11.23 | 13% |
| ایکاردی-گاتیئرز 6 | ADAR1 | 1q21.3 | 7% (1% غالب) |
| ایکاردی-گاتیئرز 7 | IFIH1 | 2q24.2 | 3% (کلاً غالب) |
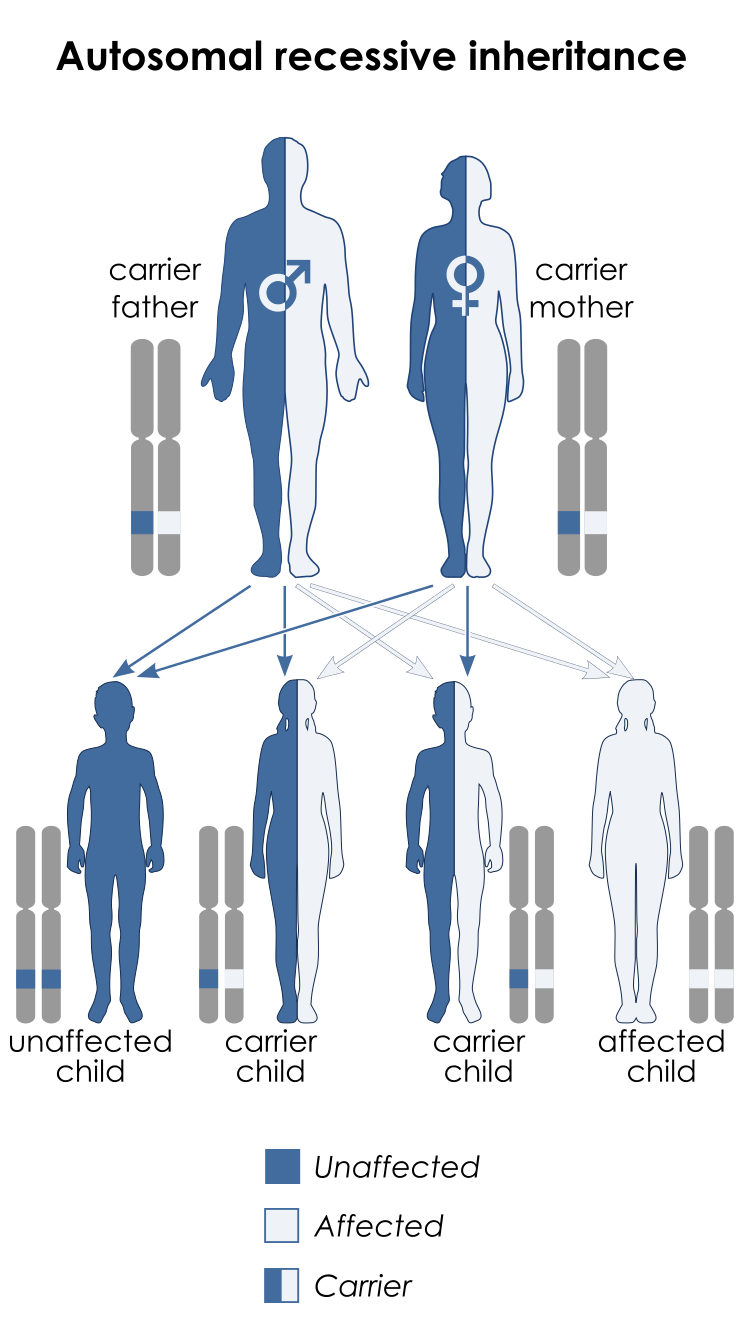
شکل 12: نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم ایکاردی-گاتیئرز در اغلب موارد از این الگو تبعیت میکند
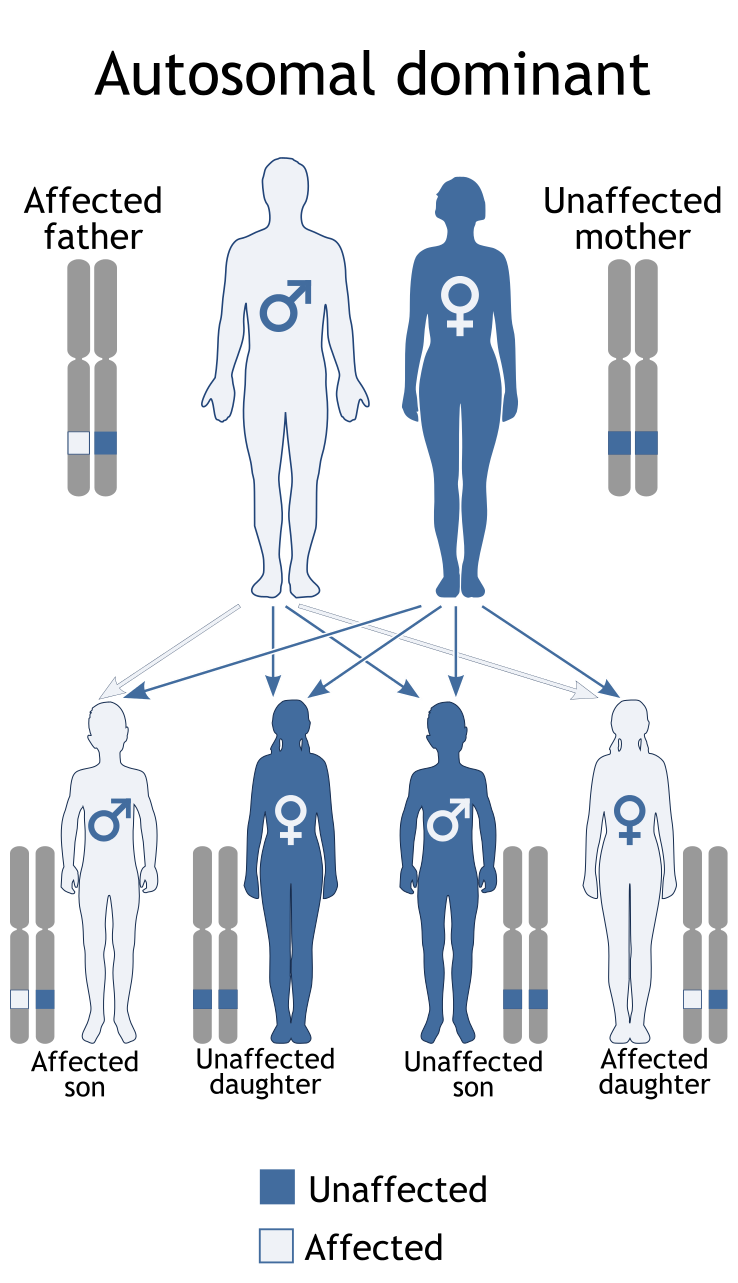
شکل 13: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم ایکاردی-گاتیئرز در برخی موارد از این الگو میتواند تبعیت کند
فراوانی سندرم ایکاردی-گاتیئرز
سندرم ای کاردی-گاتیئرز، اختلال ژنتیکی نادری است که شیوع آن در جهان بهطور دقیق مشخص نیست، با این حال، میزان فرکانس سندرم ایکاردی-گاتیئرز را میتوان از جدول شماره 1 بررسی کرد.
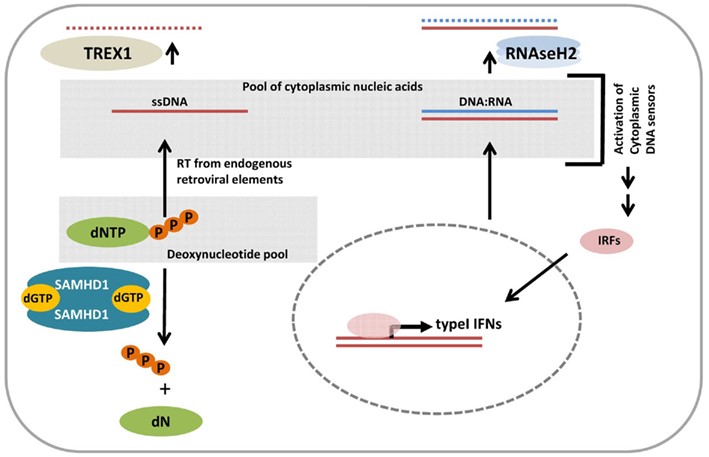
شکل 14: نمای شماتیک از مسیر مولکولی ژنهای TREX1 و SAMHD1 در مولکول DNA و RNA
تشخیص سندرم ایکاردی-گاتیئرز
سندرم ایکاردی-گاتیئرز براساس یافتههای مشخصه، ارزیابی بالینی و دقیق مبتلایان و آزمونهای تخصصی، تشخیص داده میشود. تستهای تشخیص طبی برای بررسی میزان پلاکت و آنزیمهای کبدی و کلسیم خون میتواند در تشخیص سندرم ایکاردی-گاتیئرز کمککننده باشند. همچنین تکنیکهای تصویربرداری رادیولوژی برای بررسی اندازه سر و جمجمه در تشخیص آن میتواند مؤثر باشد. دقیقترین روش تشخیص، آزمایش ژنتیک مولکولی برای ژنهای TREX1، RNASEH2A، RNASEH2B، RNASEH2C، SAMHD1،ADAR1 ،IFIH1 بهمنظور بررسی وجود جهشهای احتمالی میباشد.
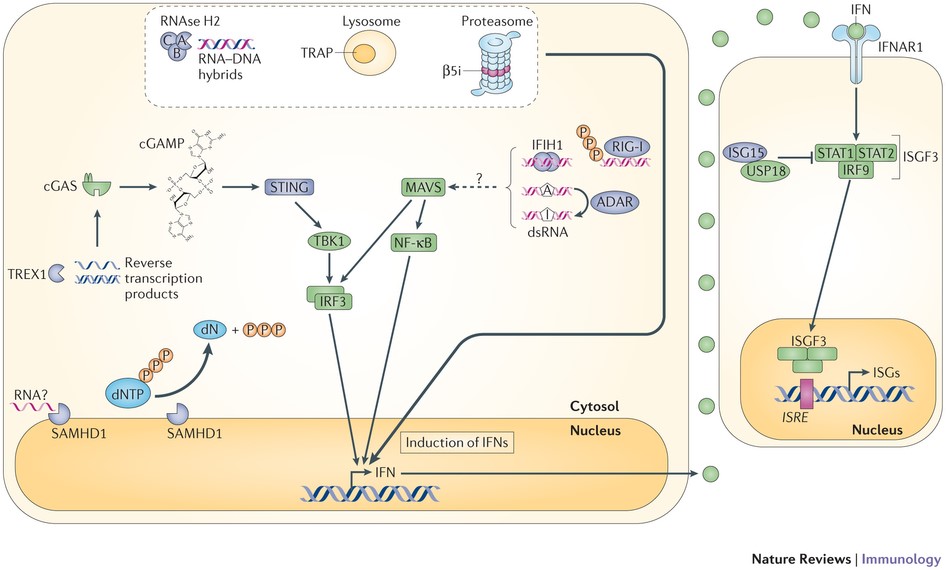
شکل 15: نمای شماتیک از مسیر مولکولی ژنهای TREX1 و SAMHD1 و IFIH1 در سیستم ایمنی بدن
مسیرهای درمانی سندرم ایکاردی-گاتیئرز
استراتژی درمان و مدیریت سندرم ایکاردی-گاتیئرز برحسب علائم و نشانههایی است که هر فرد مبتلا آن را آشکار میکند. متأسفانه برای موارد شدید این سندرم هیچ درمان استانداردی وجود ندارد و اکثر مبتلایان فرم شدید سندرم ایکاردی-گاتیئرز در همان دوران کودکی جان خود را از دست میدهند، اما در موارد خفیف این سندرم، میتوان با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص مغز و اعصاب، متخصص کبد و دستگاه گوراش، هماتولوژیست (متخصص خونشناسی)، متخصص سیستم ایمنی (ایمونولوژیست) و جراحان، به رنج مبتلایان تخفیف داد که البته همین افراد نیز عمر محدودی خواهند داشت. مشاوره ژنتیک نیز برای همه والدینی که طالب فرزندی سالم و طبیعی هستند، از جایگاه ویژهای برخوردار است.
تاریخچه سندرم ایکاردی-گاتیئرز
سندرم ایکاردی-گاتیئرز برای اولین بار در سال 1984 توسط دو پزشک فرانسوی به نامهــــای دکتر Jean Aicardi و دکتر Francoise Goutieres شناخته شده که اختلالات عصبی و کلسیفیکاسیون در مغز مربوط به این اختلال را گزارش کردند، سپس در سال 1988 دکتر Pierre Lebon و همکارانش، ویژگیهای اضافی اختلالات سیستم ایمنی مربوط به سندرم را گزارش کردند و در سال 2001 سندرم ایکاردی-گاتیئرز به رسمیت شناخته شد.

شکل 16: تصویر دکتر Jean Aicardi کاشف سندرم ایکاردی-گاتیئرز در سال 1984
منابع:
اسعدی شاهین، جمالی مهسا، باقری رعنا، توحیدیراد مانوش، سادهدل سمانه، کتاب پاتولوژی در ژنتیک پزشکی جلد اول (A-L)، صفحات 32-21، انتشارات کتب دانشگاهی عمیدی، 1396
سندرم کمخونی دیاموند- بلکفان Diamond-Blackfan Anemia Syndrome
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام