سندرم اُورباخ ویته
Urbach-Wiethe Syndrome
شاهین اسعدی (کارشناس ارشد ژنتیک)

کلیاتی از سندرم اُورباخ-ویته
سندرم اُورباخ-ویته که تحت عنوان پروتئینوز لیپوئید و یا هیالینوزیس پوست و مخاط نیز شناخته میشود، یک اختلال ژنتیکی است که با ایجاد تودههای کوچک (رسوب) پروتئینی و مولکولهای دیگر در بافتهای مختلف سراسر بدن مشخص میشود. این تودههای کوچک در پوست، دستگاه تنفسی فوقانی و بافتهای مرطوب که در قسمتهای خاص بدن مانند پلکها و داخل دهان (غشای مخاطی) و سایر مناطق هستند، ظاهر میشوند.
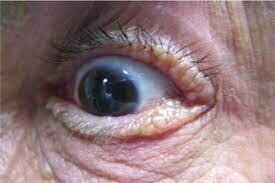
شکل 1: تصویر چشم انسان مبتلا به سندرم اُورباخ-ویته همراه با رسوب پروتئینی در بافت دهانه چشم
علائم و نشانههای بالینی سندرم اُورباخ-ویته
اولین علامت این سندرم معمولاً طنین صدای خشنی است که به علت رسوبات در تارهای صوتی حاصل میشود. در دوران کودکی، خشونت با گریه ضعیف در مبتلایان همراه است. ناهنجاریهای صوتی در طول زندگی مبتلایان سندرم اُورباخ-ویته ادامه پیدا میکند و درنهایت میتواند در صحبت کردن مشکل ایجاد کند و یا به عدم توانایی صحبت کردن منجر شود، علاوه بر این، سندرم اُورباخ-ویته میتواند گلو، لوزهها و لبها را نیز تحت تأثیر قرار دهد که منجر به مشکلات تنفسی و عفونتهای دستگاه تنفسی فوقانی خواهد شد. تودهها در غشای مخاطی ممکن است باعث ضخیم شدن و کوتاه شدن اندازه زبان گردد. این تودهها همچنین میتوانند گروهی از بافتی که زبان را به انتهای دهان متصل میکند را ضخیم کنند و توسعه زبان را دشوار سازند. ساختار زبان نیز ممکن است به دلیل آسیب به جوانههای چشایی، ظاهری صاف داشته باشد.
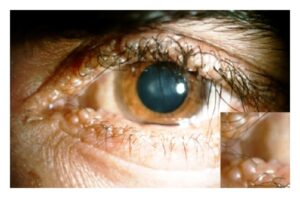
شکل 2: تصاویر اختلالات مربوط به سندرم اُورباخ-ویته
یک ویژگی مشخص از سندرم اُورباخ-ویته حضور چندین برآمدگیهای کوچک مانند دانههای تسبیح در قسمت فوقانی و تحتانی پلکها است. این برآمدگیها ممکن است باعث تحریک چشم و یا خارش شوند، اما بهطورکلی بینایی را تحت تأثیر قرار نمیدهند.
غشاهای پوستی و مخاطی اغلب در کودکان مبتلا به سندرم اُورباخ-ویته شکننده هستند که منجر به خونریزی و چروک شدن پس از ترومای جزئی میشوند. این مشکلات اغلب در ابتدای تولد، در دهان و روی پوست صورت و اندامها دیده میشود.
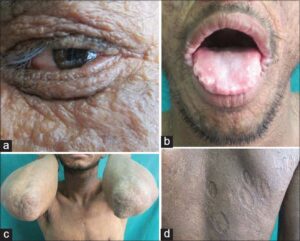
شکل 3: نمایی دیگر از اختلالات سندرم اُورباخ-ویته در پوست و مخاط
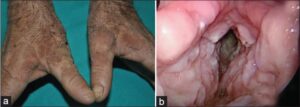
با گذشت زمان، این خراشها به شکل زخم و تاول تشکیل میشوند. تودهها در پوست انباشت میشوند که سبب پوستهپوسته شدن پوست و مایل به زرد رنگ شدن آن میشوند. آسیب پوستی بیشتر در مناطقی که اصطکاک را تجربه میکنند مانند دستها، آرنج، زانو، باسن و زیر بغل بیشتر رخ میدهد. شایان ذکر است که برخی از افراد مبتلا به سندرم اُورباخ-ویته، ریزش مو (آلوپسی) را تجربه میکنند که میتواند روی پوست سر، موهای مژهها و اَبروها تأثیر بگذارد.
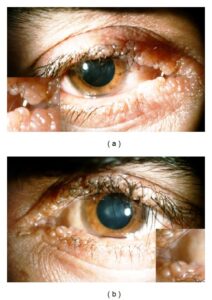
شکل 4: نمایی از ایجاد رسوب از جنس پروتئین (توده) در بافت دهانه داخلی چشم
ویژگیهای عصبی نیز در افراد مبتلا به سندرم اُورباخ-ویته رایج است؛ افراد آسیبدیده ممکن است تشنجهای مکرر (صرع) یا مشکلات رفتاری و عصبی داشته باشند که شامل سردرد، رفتار پرخاشگرانه، پارانویا، توهُم، از دست دادن حافظه کوتاهمدت و عدم ترس است. بهنظر میرسد این ویژگیها با حضور رسوب و انباشت کلسیم (کلسیفیکاسیون) در مناطقی از مغز تحت عنوان لوبهای گیجگاهی مرتبط است. لوبهای گیجگاهی به شنوایی، گفتار، حافظه و احساسات کمک میکنند. لازم به ذکر است که ناهنجاریهای مغز و ویژگیهای عصبی همیشه با یکدیگر ادغام نمیشوند، بنابراین علت ویژگیهای عصبی هنوز معلوم نیست.
این رسوبات میتوانند در برخی از اندامهای داخلی منجمله معده، بخشی از روده کوچک به نام دوازدهه و روده بزرگ یافت شوند. رسوبات در این بافتها، اغلب علائمی را ایجاد نمیکنند و در طول زمان ناپدید میشوند.

شکل 5: تصویر مرد مبتلا به سندرم اُورباخ-ویته همراه با اختلال پوستی در صورت
علتشناسی سندرم اُورباخ-ویته
سندرم اُورباخ-ویته در اثر جهش ژن ECM1 که در بازوی بلند کروموزوم شماره 1 بهصورت 1q21.2 مستقر است، ایجاد میشود. این ژن دستورالعمل لازم برای سنتز پروتئینی را فراهم میکند که در اکثر بافتها در داخل ماتریکس خارج سلولی یافت میشود و یک شبکه پیچیده است که فضاهای بین سلولی را ایجاد میکند و پوشش ساختاری را فراهم میکند. پروتئین ECM1 میتواند به پروتئینهای ساختاری متعدد (بافتها) متصل شود. علاوه بر این، پروتئین ECM1 در رشد و بلوغ سلولها، از جمله سلولهای پوستی به نام کراتینوسیتها دخیل است. پروتئین ECM1 همچنین میتواند رگزایی عروق خونی را تنظیم کند (آنژیوژنز).
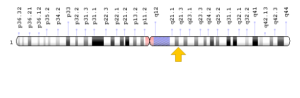 شکل 6: نمای شماتیک از کروموزوم شماره 1 که ژن ECM1 در بازوی بلند این کروموزوم بهصورت 1q21.2 مستقر است
شکل 6: نمای شماتیک از کروموزوم شماره 1 که ژن ECM1 در بازوی بلند این کروموزوم بهصورت 1q21.2 مستقر است
جهشهای ژن ECM1 که باعث سندرم اُورباخ-ویته میشوند، باعث تولید یک پروتئین غیرفعال یا عدم تولید پروتئین میشود. کمبود پروتئین ECM1 عملکرد اتصال این پروتئین با پروتئینهای دیگر را کاهش میدهد که همین امر منجر به ایجاد ماتریکس خارج سلولی بیثبات (ناپایدار) میشود. بدون حمایت کافی از ماتریکس خارج سلولی، سلولهای پوست و دیگر بافتها تضعیف میشوند، بااینحال علت رسوب (تودهها) در پوست و دیگر بافتها مشخص نیست. ماتریکس خارج سلولی ناپایدار ممکن است باعث ایجاد سلولهای ذخیرهکننده پروتئینوز لیپوئید شوند.
سندرم اُورباخ-ویته از الگوی توارثی اتوزومال مغلوب پیروی میکند، بنابراین برای ایجاد این سندرم، دو نسخه از ژن جهشیافته ECM1 (یکی از پدر و دیگری از مادر) موردنیاز است و شانس داشتن فرزند مبتلا به سندرم اُورباخ-ویته در حالت اتوزومی مغلوب، برای هر بارداری احتمالی به میزان 25% است.
فراوانی سندرم اُورباخ-ویته
سندرم اُورباخ-ویته اختلال ژنتیکی نادر است که تاکنون کمتر از 500 مورد مبتلا به این سندرم از سراسر جهان در ادبیات پزشکی گزارش شده است. این سندرم در کشورهای ایران، ترکیه و آفریقای جنوبی بیشتر رخ میدهد.
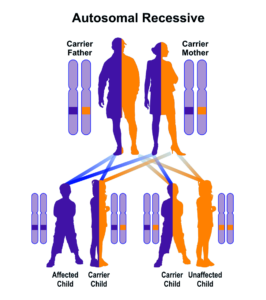
شکل 7: نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم اُورباخ-ویته نیز از این الگو تبعیت میکند
تشخیص سندرم اُورباخ-ویته
سندرم اُورباخ-ویته بر اساس یافتههای بالینی و فیزیکی مبتلایان و برخی آزمایشهای پاتولوژیکی، تشخیص داده میشود. دقیقترین روش تشخیص این سندرم، آزمایش ژنتیک مولکولی برای ژن ECM1 بهمنظور بررسی وجود جهشهای احتمالی است. تشخیص پیش از تولد نیز با استفاده از تکنیک PGD و مایع آمنیوسنتز و یا نمونهبرداری از پرزهای کوریونی جفت جنین امکانپذیر است. روشهای رنگآمیزی PAS و همچنین روشهای ایمونوهیستوشیمی نیز در تشخیص این سندرم بکار میروند.
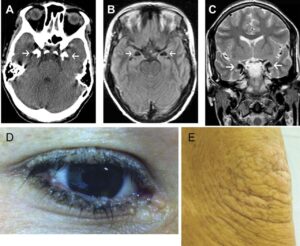
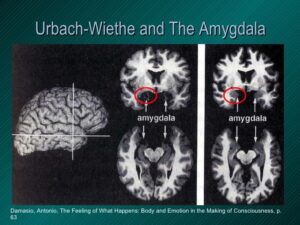
شکل 8: تصاویر رادیولوژی از اختلال مغزی در سندرم اُورباخ-ویته
مسیرهای درمانی سندرم اُورباخ-ویته
استراتژی درمان و مدیریت سندرم اُورباخ-ویته بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص پوست و مو، متخصص دهان و متخصص حنجره و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. در حال حاضر، هیچ درمان قاطعی برای این سندرم وجود ندارد و تمامی اقدامات بالینی بهمنظور تخفیف رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که خواستار فرزندی سالم هستند، از اهمیت ویژهای برخوردار است.
تاریخچه سندرم اُورباخ-ویته
این سندرم برای اولین بار در سال 1908 توسط دکتر Friedrich Siebenmann پزشک متخصص گوش و حلق و بینی در دانشگاه بازل سوئیس گزارش گردید. سپس در سال 1925 یک پزشک متخصص پوست اهل کشور سوئیس به نام Friedrich Miescher موارد بیشتری از این سندرم را توصیف کرد و سرانجام در سال 1929 توسط دکتر Erich Urbach پزشک متخصص پوست و دکتر Camilo Wiethe پزشک متخصص گوش و حلق و بینی اهل شهر وین اُتریش، این سندرم بهطور کامل گزارش گردید و به رسمیت شناخته شد.

شکل 9: تصویر دکتر Erich Urbach یکی از کاشفان سندرم اُورباخ-ویته
منبع:
اسعدی شاهین، ملتیار حسن، روشنروان ندا، جمالی مهسا، داداشپور مهدی، محمدزاده حمیده، فتاحی محیا، کتاب پاتولوژی در ژنتیک پزشکی جلد 6 (M-Y)، صفحات 876-865، انتشارات کتب دانشگاهی عمیدی، بهار 1397
https://www.sciencedirect.com/science/article/abs/pii/S187140480500002X
https://medlabnews.ir/%d8%b3%d9%86%d8%af%d8%b1%d9%85-%d9%be%d9%8f%d9%85%d9%be/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام