ارزیابی کیفیت خارجی؛ مروری بر طراحی، تواناییها و کاستیهای برنامههای کنونی
دكتر حسن بيات دكتراي علوم آزمايشگاهي
این نوشته به پیشنهاد جناب آقای دکتر حسن هاشمی مدنی، مدیر محترم برنامهی ارزیابی کیفیت خارجی انجمن دکترای علوم آزمایشگاهی (EQAP) به این قصد که کمک کوچکی باشد در راستای تلاشهای گستردهی دستاندرکاران محترم EQAP برای ارتقاء کیفیت آن برنامه؛ و با نظر لطف و قول مساعد جناب آقای دکتر محمدرضا عابدی سردبیر محترم ماهنامهی اخبار آزمایشگاهی برای چاپ در این ماهنامهی وزین نگاشته شده است. امیدوارم مطالعهی آن برای جامعهی آزمایشگاهی کشورمان خالی از بهره نباشد.
حسن بیات؛ دانشآموختهی علوم آزمایشگاهی – اردیبهشت 1393
مقدمه:
هدف از ارزیابی کیفیت خارجی (EQA[1])، یا آزمون مهارت (PT[2])، آن است که بتوان به طور پیوسته تایید کرد که نتایج تولید شده به وسیلهی آزمایشگاهها با کیفیتی که برای مراقبت از بیماران لازم است مطابقت دارد. فعالیتهای EQA بیش از 60 سال پیش و در واکنش به مشاهداتی که نشان میداد وقتی یک نمونه تقسیم و به آزمایشگاههای گوناگون فرستاده میشد نتایج متفاوتی به دست میآمد، به عنوان ابزاری آموزشی پای به عرصهی آزمایشگاه پزشکی گذاشت. در آن روزگار، روشهای سنجش به وسیلهی آزمایشگاهها ساخته میشدند و از نظر جزئیات و کالیبراسیون با هم تفاوت داشتند. بنا بر این، از نتایج EQA به عنوان محرکی برای عیارمندسازی روشها و کالیبراتورها بین آزمایشگاههای مختلف استفاده میشد. از آن زمان تا کنون، برنامههای EQA از نظر وسعت و پیچیدگی بسیار متحول شدهاند و در حال حاضر جزئی اساسی از سامانهی مدیریت کیفیت آزمایشگاه هستند.
تا حدود یک دهه پیش، بیشتر فعالیتهای مدیریت کیفیت بر کاهش نوسان درونآزمایشگاهی و ارزیابی نوسان بینآزمایشگاهی متمرکز بود. در سالهای اخیر اهمیت کاهش عدمصحت، در هر دو شکل درونآزمایشگاهی و بینآزمایشگاهی، آشکار شده است. بیماران امروزه بیشتر به وسیلهی یک گروه از پزشکان معالجه میشوند تا یک پزشک واحد، و در روند درمان به مراکز درمانی متعدد مراجعه و جوابهای آزمایشگاهی از چند آزمایشگاه مختلف دریافت میکنند. بنا بر این، حذف عدمصحت و به حداقل رسانیدن عدمدقت در فرآیند رسیدگی به بیماران امری بسیار اساسی است. حتا عدمصحت جزئی روشها میتواند بر طبقهبندی درست بیمار و شمار بیمارانی که مراقبت نامناسب دریافت میکنند تاثیر بزرگی داشته باشد؛ به ویژه برای آزمایشهایی که برای آنها از مقادیر برشگاهی یکسان استفاده میشود. به عنوان نمونه میتوان به سنجش لیپیدها و لیپوپروتئینها اشاره کرد که برای آنها در سراسر جهان از مقادیر برشگاهی یکسانی در پیشگیری و درمان بیماریهای قلبی-عروقی استفاده میشود.
در سالهای اخیر در راستای کاستن از عدمصحت نتایج و ایجاد هماهنگی بین نتایج آزمایشگاههای مختلف، تلاشهای فراوانی به وسیلهی سازمانهایی مانند IFCC و AACC آغاز شده است. این فعالیتها به دو گروه عیارمندسازی[3] و هماهنگسازی[4] تقسیم میشوند. چنانچه یک آنالیت به طور مشخص تعریف شده باشد و برای سنجش آن آنالیت روش مرجع و معیار[5] مرجع وجود داشته باشد، اقدامات مربوط به یکدستکردن نتایج حاصل از سنجش آن آنالیت با روشهای گوناگون “عیارمندسازی” نامیده میشود. اما چنانچه برای یک آنالیت هر یک از این سه عامل یعنی تعریف مشخص، روش مرجع و معیار مرجع فراهم نباشد، اقدامات مربوط به یکسان سازی نتایج حاصل از روشهای مختلف “هماهنگسازی” نامیده میشود. در حال حاضر برنامههای EQA جایگاه بسیار مهمی در فعالیتهای عیارمندسازی/هماهنگسازی دارند.
عامل اصلی در تفسیر نتایج EQA عبارت است آگاهی در بارهی چگونگی تبادلپذیری مواد کنترل و فرآیند به کار گرفته شده برای تعیین مقدار هدف. یک مادهی EQA تبادلپذیر مادهای است که وقتی آن را با روشهای گوناگون اندازهگیری میکنیم، رابطهی عددی بین نتایج حاصل از این روشها همسان است با رابطهای که از سنجش نمونههای بیماران با آن روشها به دست میآید. نمونههای EQA تبادلناپذیر، دارای اختلاف وابسته به زمینه با مقدار نامعین هستند که سبب میشود تفسیر نتایج حاصل از آنها با محدودیت روبرو شود. از نمونههای EQA تبادلپذیر میتوان برای ارزیابی صحت در مقابل یک روش مرجع یا یک روش مقایسهای منتخب استفاده کرد. به علاوه، چگونگی همخوانی دیده شده در سنجش یک مادهی تبادلپذیر با روشهای گوناگون، بازتابی است از چگونگی همخوانییی که در سنجش نمونههای بیماران با آن روشها دیده خواهد شد. برای بررسی نتایج EQA حاصل از نمونههای تبادلناپذیر باید ارزیابی گروهی انجام داد یعنی شرکتکنندگان را بر اساس روش سنجش گروهبندی کرد و نتیجهی شرکتکننده را با میانگین/میانهی گروه مقایسه کرد؛ با این فرض که روشهای تشکیل دهندهی آن گروه دارای اختلاف وابسته به زمینهی یکسان یا خیلی نزدیک به هم هستند. هدف از ارزیابی گروهی بررسی این است که آیا عملکرد یک آزمایشگاه در به کاربستن یک روش، با ویژگیهای سازندهی آن روش و/ یا عملکرد دیگر آزمایشگاههایی که آن فناوری را دارند همخوانی دارد یا نه. نمونههای EQA تبادلناپذیر اطلاعات معناداری در بارهی ارتباط بین نتایج بیماران که از روشهای گوناگون حاصل میشوند فراهم نمیکنند.
در این نوشته جنبههای کلیدی در طراحی، اجرا و تفسیر برنامههای EQA، همراه با نمونههایی از برنامههای EQA در کشورهای پیشتاز در این زمینه ارائه میشود و به بیان تواناییها و کاستیهای شیوههای گوناگون EQA و همچنین توضیح این امر که چگونه برنامههای EQA میتوانند در پیشرفت عرصهی آزمایشگاه سهیم باشند میپردازد. هدف این نوشتار آن است که بتواند معیاری فراهم کند برای مقایسهی برنامههای کنونی EQA در کشور خودمان اگرچه به بحث و نتیجهگیری در بارهی برنامههای جاری در ایران پرداخته نخواهد شد. این مرور محدود خواهد بود به برنامههای EQA برای روشهای کمی.
اصول کلی
ضرورت اساسی یک برنامهی EQA این است بتواند اطمینان شرکتکنندگان را نه تنها به اجرای برنامه بلکه همچنین به ارزشمندی علمی آن برنامه جلب و حفظ کند و گرنه شرکتکنندگان پس از دریافت گزارش ارزیابی، اقدامی انجام نخواهند داد و آن برنامه محرکی برای پیشرفت نخواهد بود. برای نیل به چنین مقصودی، پیشنیازهای بنیادین زیر لازم است:
- سرعت کافی در آگاهسازی. قابل دستیابی از راه:
- توزیع مکرر.
- برگرداندن سریع گزارش بررسی نتایج.
- تبادل موثر اطلاعات مربوط به عملکرد. قابل دستیابی از راه:
- گزارشهای ساختارمند، دربردارندهی اطلاعات کافی، و آسان فهم.
- سامانهی امتیازدهی انباشتی.
- مبنای مناسب برای ارزیابی. قابل دستیابی از راه:
- نمونههای پایدار و همگن که شبیه نمونههای بالینی رفتار میکنند.
- مقادیر هدف قابل اعتماد و ارزشمند.
رعایت موارد بالا برای این که یک برنامه بتواند از طریق افزایش همسانی بین نتایج آزمایشگاههای مختلف، ویژگی اعتمادپذیری نتایج بیماران را افزایش دهد ضروری است. فرآیند سنجش نمونهها در آزمایشگاه، گزارش نتایج به برگزارکننده و برگشت گزارش ارزیابی به آزمایشگاهها باید پیوسته و سریع باشد تا این امکان را برای آزمایشگاه فراهم کند که بتواند کاستیهای گزارش شده را بررسی و اصلاح کند. سامانهی امتیازدهی باید از استحکام علمی و اعتمادپذیری برخوردار باشد و مستقل از عملکرد دیگر شرکتکنندگان باشد تا بتوان بر اساس آن، هم عملکرد هر آزمایشگاه منفرد را و هم عملکرد کلی هر روش را در طول زمان بررسی و در بارهی عملکرد بلندمدت آنها اطلاعات فراهم کرد. نمونههای توزیع شده باید مناسب برای کاربرد مورد نظر باشند. عواملی که باید در نظر گرفت عبارت است از منشا نمونهها، افزودنیها یا نگهدارندهها، شکل نمونه (مایع، یخزده، یا لیوفیلیزه) و مهمتر از اینها پایداری نمونه، تبادلپذیری و نبود دیگر اثرات وابسته به زمینه که ممکن است مانع ارزیابی درست روشها شود. مقادیر هدف نمونههای فرستاده شده باید قابل اعتماد باشد و دست آخر این که گزارشهای ارائه شده به آزمایشگاه باید در حالی که اطلاعات کافی در بارهی عملکرد آزمایشگاه و شیوههای گوناگون سنجش را در بر دارند در همان حال به آسانی قابل فهم باشند.
تعداد نمونههای فرستاده شده در برنامههای گوناگون متفاوت است و گسترهی وسیعی را شامل میشود؛ از حداقل ممکن شامل فقط دو توزیع در سال با دو نمونه در هر توزیع تا 12 توزیع در سال با 5 نمونه در هر توزیع (60 نمونه در سال برای هر آنالیت). تعداد مناسب توزیع و نیز تعداد مناسب نمونه در هر توزیع به عوامل چندی بستگی دارد شامل پیچیدگی ساختار آنالیت موردنظر، بلوغ فناوری سنجش و مقصود از اجرای برنامه. برای مثال برای آنالیتهایی که ساختار همگنی دارند و فناوری سنجش آنها به خوبی توسعه یافته و قابل اعتماد است برنامههای نسبتاً ساده با توزیع تعداد کمی نمونه ممکن است مناسب باشد. در مورد این آنالیتها کافی است که نشان دهیم نتایج شرکتکنندگان برای برآورده کردن الزامات قانونی، به اندازهی کافی به مقدار هدف نزدیک است. اما برای آنالیتهایی که ساختاری ناهمگن دارند و در روشهای ایمونولوژیک مختلف به طور متغیری شناسایی میشوند برنامههای کاملتر و وسیعتری لازم است.
نمونههای EQA
نمونههای ایدهآل EQA باید با مجموعهای از معیارها مطابقت داشته باشند: پایداری در شرایط انتقال و نگهداری، همگنی بین همهی قسمتهای توزیع شده، در بر داشتن غلظتهای مناسب برای گسترهی بالینی موردنظر، مناسب بودن نوع نمونه (ادرار، خون کامل، سرم)، در دسترس بودن در حجم کافی، ارزان بودن طوری که قیمت آن مانعی برای اجرای برنامه نباشد و داشتن رفتاری همانند با نمونههای بیمار در روشهای گوناگون. در عمل، برآورده کردن همهی این الزامات ممکن نیست و بسته به مورد باید سازشهایی به عمل آید. تبادلپذیری با نمونههای بیماران یکی از مهمترین مفاهیمی است که بر طراحی و تفسیر برنامههای EQA تاثیرگذار است.
تبادلپذیری[6]
بر اساس تعریف ISO/REMCO N1129، تبادلپذیری ویژگییی از یک مادهی مرجع یا EQA است که به سبب آن رابطهی عددی بین نتایج سنجش آن مادهی مرجع یا EQA با روشهای گوناگون، همسان است با رابطهی حاصل از استفاده از آن روشها در سنجش یک مجموعه نمونه که نمایندهی نمونههای بالینی بیماران است. به بیان ساده یک مادهی مرجع یا EQA را وقتی تبادلپذیر میدانیم که رفتار آن در روشهای مختلف سنجش، نظیر رفتار نمونههای معمولی بیماران در آن روشها باشد و بنا بر این بتوان از اطلاعات به دست آمده از سنجش آن ماده با روشهای گوناگون استفاده کرد برای داوری و اقدام در بارهی وضعیت سنجش نمونههای بیماران با آن روشها؛ و این یعنی میتوان بین نتایج آن ماده و نتایج نمونههای بیماران تبادل اطلاعات کرد. شکل 1 مثالی از بررسی تبادلپذیری چند ماده EQA برای دو روش اندازهگیری را نشان میدهد که در آن از سنجش مجموعهای از نمونههای منفرد بیماران استفاده شده است. در این مثال با استفاده از واکاوی رگرسیون، رابطهی عددی بین نتایج بیماران تعیین شده است و فاصلهی پیشبینی 95% در دو طرف خط رگرسیون که با استفاده از انحراف معیار باقیماندهها (SDxy) تعیین شده است محدودهای است که انتظار میرود نتایج حاصل از سنجش مادهی EQA، در صورت تبادلپذیر بودن، در آن ناحیه قرار بگیرد. همانطور که در این شکل دیده میشود از 7 مادهی EQA بررسی شده، 6 مورد تبادلپذیر هستند و یک مورد (مادهی F) تبادلناپذیر است.
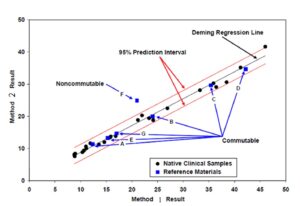
شکل 1 – مثالی از بررسی ویژگی تبادلپذیری نمونههای EQA با استفاده از نمونههای منفرد بیماران
خط رگرسیون که از میان نتایج عبور میکند نمایانگر رابطهی ریاضی موجود بین این دو روش سنجش در سنجش نمونههای بیماران است. ناحیهی پیشبینی 95% ناحیهای است که نتایج نمونههای EQA تبادلپذیر در آن قرار میگیرد.
نتیجهی حاصل از یک مادهی EQA تبادلپذیر برابر است با نتیجهای که از سنجش یک نمونهی بیمار با همان مقدار آنالیت انتظار میرود. یک مادهی EQA که برای روشهای گوناگون تبادلناپذیر است نمیتواند اطلاعات سودمندی در بارهی رابطهی موجود بین نتایج اندازهگیری نمونههای بیماران با آن روشها فراهم کند.
اصطلاح “اختلاف وابسته به زمینه” و “اثر زمینهای” برای اشاره به اختلاف ناشی از تبادلناپذیری به کار برده میشوند. در مبحث EQA، منظور از اصطلاحات تبادلناپذیری، اختلاف وابسته به زمینه و اثر زمینهای، تفاوتی است که تنها در نمونههای EQA رخ میدهد اما در نمونههای اصلی بیماران بالینی دیده نمیشود. در نتیجه، در EQA تداخل ناشی از یک مادهی درونزاد (مثلا بیلیروبین) عموما به عنوان اثر زمینهای در نظر گرفته نمیشود، اما تبادلناپذیری ناشی از یک آنالیت غیربومی (دارای منشاء غیر انسانی) مثلا دیتائورو بیلیروبین اثر زمینهای به شمار میآید.
تهیهی نمونههای احتمالاً تبادلپذیر
برای تهیهی نمونههای تبادلپذیر، باید نمونه را به روش همسان با نمونههای بیماران جمعآوری و پردازش کرد وسپس در شرایط پایدار، تقسیم و پخش کرد. میتوان از نمونههای اهدائی به شکل منفرد یا انباشته استفاده کرد. معمولاً حجم مورد نیاز و یا غلظت/فعالیت مورد نظر، استفاده از نمونههای منفرد را محدود میکند. از دیگر کاستیهای نمونههای منفرد، احتمال وجود مداخلهگری در نمونه است که ممکن است تنها بر تعدادی از روشها تاثیرگذار باشد. در نمونههای انباشته شده مداخلهگر موجود در یک نمونه رقیق میشود و بسته به تعداد نمونههای رویهم ریخته شده، ممکن است اثر آن حذف شود. با وجود این، یک محدودیت قابل توجه نمونههای انباشته این است که امکان دارد به دلیل واکنش بین اجزای نمونههای گوناگون، مانند پروتئینهای سرم یا کمپلکسهای ادرار، تجمع یا آگلوتیناسیون رخ دهد و در نتیجه پردازشهای اضافی لازم افتد که خود میتواند سبب تغییر زمینه شود.
روشهای به کارگرفته شده برای جمعآوری و پردازش نمونهها، عوامل مهمی در جلوگیری از تغییر زمینه و حفظ تبادلپذیری در فرآوردهی نهایی هستند. راهکارنمای CLSI[7]-C37A دستورکار مستحمکی است که برای تهیهی نمونههای تبادلپذیر برای سنجش کلسترول نوشته شده است. در این دستورکار مراحل جمعآوری خون، تهیهی سرم، تهیهی انباشته و فریزکردن قسمتها در شرایطی که ویژگیهای تبادلپذیری کلسترول را تغییر نمیدهد شرح داده شده است. اگرچه این دستورکار در اصل برای نمونههای کلسترول نوشته شده است اما کارآمدی آن در تهیهی نمونههای تبادلپذیر برای تریگلیسریدها، HDLc و کراتینین نیز ارزشیابی شده است. و نیز به رغم این که قابلیت استفاده از C37A در تهیهی نمونههای تبادلپذیر برای آنالیتهای دیگر ارزشیابی نشده است اما در حال حاضر بهترین شیوهی موجود است و در چندین برنامهی بررسی صحت روشهای سنجش آنالیتهای گوناگون، از این دستورکار برای تهیهی مواد تبادلپذیر از نمونههای منفرد یا انباشته استفاده شده و نتایج رضایتبخشی به دنبال داشته است. برای زمینههای دیگر غیر از سرم، دستورکارهای مستحکمی ارائه نشده است اما اصول عمومی شامل جمعآوری نمونههای تغییر نیافته، انباشتن، پخش کردن و اندازهگیری بیدرنگ آنها یا فریزکردن قسمتها در دمای ≤ 70o C بهترین شیوهای که برای تهیهی نمونههای EQA که احتمالا تبادلپذیر خواهند بود در دسترس است.
یکی از محدودیتهای نمونههای اهدایی این است که ممکن است غلظت یا فعالیت موردنظر در دسترس نباشد. در این صورت باید غلظتهای بالاتر را با افزودن آنالیت به انباشتهی تغییرنیافته تهیه کرد. میتوان چنین فرض کرد که افزودن آنالیت خالص، زمینه را تغییر نمیدهد و تبادلپذیری باقی خواهد ماند. درستی چنین فرضیهای برای افزودن کراتینین به انباشتهی سرمی گزارش شده است. با وجود این، چنین فرضی برای آنالیتهای ساده معقول است. با پیچیده شدن ساختار یا کاسته شدن از خلوص آنالیت، یا چنانچه زمینهی مادهی افزوده شده در تغییر زمینهی مادهی بومی نقش داشته باشد، اعتماد به چنین فرضیهای سست میشود. غلظتهای پائینتر را میتوان با برداشت آنالیت، مثلا با جذب ایمنی بر یک فاز جامد، تهیه کرد. البته برداشت آنالیت ممکن است به برداشت ناخواستهی مولکولها و به عبارت دیگر به تغییر زمینه بینجامد؛ به ویژه هنگامی که از تکنیکهای غیراختصاصی مانند ذغال یا پروتئین A استفاده میشود.
ارزشیابی تبادلپذیری نمونهها
دستورکار توافقی CLSI-EP30A (که قبلا C53A نامیده میشد) برای ارزشیابی تبادلپذیری مواد EQA نوشته شده است. بهترین حالت آن است که در یک برنامهی EQA در هر دور توزیع نمونهها، تبادلپذیری مواد تهیهشده را با استفاه از نمونههای تک-دهنده ارزشیابی کرد. با وجود این، ممکن است تهیهی چنین نمونههایی مشکل باشد و نیز چنین ارزشیابییی هزینهبر است. بنا به دلایل عملی، ممکن است تبادلپذیری نمونهها را فقط در یک توزیع ارزشیابی کرد و چنانچه تایید شد، میتوان ساختهای بعدی را که به همان شکل تهیه میشوند تبادلپذیر فرض کرد. البته باید توجه داشت که در صورت افزودهشدن روشهای جدید به برنامه، باید تبادلپذیری را برای روشهای جدید ارزشیابی کرد.
در حال حاضر، معمولا بر اساس رعایت سختگیرانهی اصولی که در بالا آمد، فرض بر آن گذاشته میشود که مواد تهیه شده تبادلپذیر هستند. اگرچه این فرض معقول است اما احتمال تبادلناپذیری محدودیتی است که در تفسیر نتایج باقی میماند. هرچه پردازش نمونهها از آن چه که برای نمونههای بیماران انجام میشود متفاوتتر باشد، فرض تبادلپذیری سستتر میشود.
تهیهی نمونههای احتمالاً تبادلناپذیر
کارخانههای سازندهی مواد EQA شیوههای گوناگون و عموماً انحصاری را برای تهیهی مواد دارای غلظت مناسب و نیز ویژگیهای پایداری و نگهداری مطلوب به کار میبندند. مواد EQA در طول فرآیند تهیه به کرات دستخوش تغییرات گوناگونی میشوند که به تبادلناپذیری آنها میانجامد. در شکل 2 شیوهی کلی تهیهی آنچه که در برنامههای EQA “سرم” نامیده میشود نمایش داده شده است که نمونهای است از برخی تاثیرات مهم بر زمینه که طی فرآیند ساخت رخ میدهد و ممکن است بر ویژگیهای تبادلپذیری مادهی نهایی تاثیرگذار باشد. تبادلناپذیری نسبت داده میشود به عواملی شامل تغییر زمینه حتا اگر آن نمونه از منابع انسانی سرچشمه گرفته یا تهیه شده باشد، اشکال غیربومی یک آنالیت که هنگام سنجش پیغامی متفاوت با پیغام حاصل از آنالیت بومی تولید میکنند، ناخالصیهای ناشی از افزودههای آنالیتی، فرآیند محافظت و دیگر تاثیراتی که در نمونههای بالینی بومی وجود ندارد.
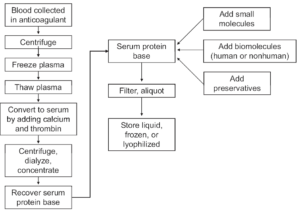
شکل 2 – مراحل نمادین تهیهی نمونههای سرمی EQA که میتواند تاثیرات احتمالی بر تبادلپذیری محصول داشته باشد.
مقدار هدف و معیارهای پذیرش نتایج EQA
برای این که بتوان نتایج EQA را تفسیر کرد باید سازماندهندگان برنامه برای هر آنالیت یک مقدار هدف و یک بازهی نتایج قابلقبول اطراف آن هدف تعیین کنند. چون هدف اصلی برنامههای EQA ارزیابی عدمصحت است، یعنی تعیین این که نتایج آزمایشگاهها چقدر با “مقدار درست” اختلاف دارند، بنا بر این باید مقدار هدف نمایندهی مقدار درست باشد. این مهم است که سازماندهندگان برنامههای EQA، شرکتکنندگان را از شیوههای گوناگون به کار گرفته شده در فرآیند تعیین هدف و نقاط قوت و ضعف آن شیوهها آگاه نمایند؛ چرا که تنها در این صورت است که شرکتکنندگان میتوانند بر اساس گزارشهای EQA اقدام درست را انجام دهند.
بهترین حالت آن است که همهی مقادیر هدف به طور کامل ارزشیابی شوند یعنی با استفاده از روشهای مرجع یا روشهای مقایسهای منتخب تعیین مقدار شوند، اگرچه چنین کاری در عمل بسیار سخت است. به عنوان راهکاری جایگزین میتوان نتیجهی حاصل از سنجش مادهی کنترل در یک یا چند آزمایشگاه مرجع را به عنوان مقدار هدف در نظر گرفت به شرط آن که عمکلرد آن آزمایشگاه(ها) به طور چشمگیری بهتر از دیگران باشد؛ امری که به باور نویسندگان راهکارنمای WHO برای EQA “در بیشتر موارد چنین نیست یا قابل اثبات نیست”. احتمال وارد کردن خطاهایی که ناشی از عدمصحت خود این آزمایشگاهها است و نیز ایجاد حس اعتماد به نفس زیادی برای ایشان، خطر ذاتی این شیوهی تعیین هدف است. راهکار دیگر استفاده از میانگین یا میانهی شرکتکنندگان با عنوان “مقدار هدف توافقی” است. هیچ مبنای علمییی برای این که این هدفهای توافقی درست باشند وجود ندارد، اما تجربیات عملی نشان میدهد غالباً میانگین تعداد زیادی شرکتکننده به عنوان مقدار هدف به اندازهی کافی قابل اطمینان است. با وجود این، ارزشمندی آنها را نباید مفروض انگاشت بلکه باید آن را از طریق بررسی تکرارپذیری، بازیافت و ارزیابیهای مقایسهای با دیگر برنامههای EQA اثبات کرد.
ارزشگذاری مقدار هدف وقتی که نمونهها تبادلپذیر هستند
یک مزیت کلیدی استفاده از نمونههای تبادلپذیر فراهم شدن امکان ارزیابی ردیابیپذیری[8] نتایج به یک سامانهی مرجع است. برای نیل به این مقصود باید از یک روش مرجع یا از یک روش مقایسهای بسیار اختصاصی که خود به یک روش مرجع ردیابیپذیر است برای ارزشگذاری مادهی کنترل استفاده شود. در صورت در دسترس بودن، باید ردیابیپذیری به روشها، مواد و آزمایشگاههایی باشد که در پایگاه دادههای JCTLM[9] فهرست شدهاند (http://www.bipm.org/jctlm/). همچنین میتوان از انتقال ارزش بر اساس نتیجهی سنجش یک مادهی مرجع گواهیشده (CRM[10]) استفاده کرد به شرط آن که تبادلپذیری آن مادهی مرجع تایید شده باشد. در مورد نمونههایی که از افزودن آنالیت به یک نمونهی عاری از آن آنالیت تهیه میشوند، ارزشگذاری مادهی کنترل بر اساس وزن مادهی افزوده شده بستگی دارد به خلوص مادهی افزوده شده، درستی ابزار اندازهگیری و همسانی دیده شده بین شکل خالصشده و شکل بومی آن آنالیت در نمونههای انسانی. توصیه میشود که تبادلپذیری نمونههای نهایی که از افزودن آنالیت به دست میآیند پس از تهیه ارزشیابی شود. به عنوان آخرین راهکار و وقتی که سامانهی مرجعی برای اندازهگیری یک آنالیت در دسترس نباشد، میتوان از میانگین یا میانهی همهی شرکتکنندگان پس از حذف دادههای پرت به عنوان مقدار هدف استفاده کرد زیرا در مورد یک مادهی تبادلپذیر انتظار میرود همهی روشها نتایج یکسانی تولید کنند.
ارزشگذاری وقتی که تبادلپذیری نمونهها محتمل نیست
در مورد این نمونهها، رایجترین فرآیند برای تعیین هدف عبارت است از دستهبندی روشها به گروههایی که هر کدام نمایندهی فناوری یکسانی هستند و پس از حذف دادههای پرت در نظر گرفتن میانگین یا میانهی هر گروه به عنوان هدف. یک “گروه” تشکیل میشود از روشهایی که احتمالا برای یک مادهی معین EQA اختلاف وابسته به زمینهی یکسانی دارند و بنا بر این انتظار میرود که آن روشها نتایج یکسانی برای آن مادهی EQA تولید کنند. روش معمول این است که گروهها بر اساس گروهبندی دستگاه/معرف تشکیل میشوند. یکی از محدودیتها هنگام محاسبهی میانگین یا میانه برای گروهها، تعداد شرکتکنندگان است. به موازات کاهش تعداد نتایج یا افزایش پراکندگی نتایج، عدم قطعیت مقدار هدف افزایش مییابد. چنانچه پراکندگی نتایج محدود باشد، مقایسهی نتایج با میانه میتواند ارزیابی سودمندی را فراهم سازد.
وقتی که تبادلپذیری نمونهها مشخص نیست، ارزشگذاری نمونه با یک روش مرجع سود چندانی ندارد زیرا امکان ندارد مشخص کنیم که آیا انحراف نتیجهی شرکتکننده از مقدار هدف، به دلیل اختلاف کالیبراسیون آن روش است یا این که نتیجهی اختلاف وابسته به زمینه است. هدف تعیین شده با روش مرجع برای مواد تبادلناپذیر، در ارزیابی روشهای اختصاصیت-بالا سودمندی بیشتری دارد و برعکس برای روشهای اختصاصیت-پایین چندان سودمند نیست. به همین شکل، میانگین/میانهی حاصل از همهی روشها نیز چندان مفید نیست مگر آن که شواهدی در دست باشد که از همسانی اختلاف وابسته به زمینه در مورد همهی روشها پشتیبانی کند و یا این که جایگزین دیگری نباشد (مثلا اندازهی همهی گروهها خیلی کوچک باشد). در صورت استفاده از میانگین/میانهی همهی روشها، گروههای بزرگتر تاثیر بیشتری بر مقدار هدف خواهند داشت و نیز بسته به تعداد نسبی شرکتکنندگانی که از روشهای گوناگون استفاده میکنند ممکن است مقدار هدف در طول زمان متغیر باشد و ممکن است آن مقدار هدف دست کم برای بررسی تعدادی از گروهها مناسب نباشد. متغیر بودن مقدار هدف این مشکل را ایجاد میکند که نمیتوان نتایج اخیر را با نتایچ پیشین مقایسه و تغییرات عملکرد در طول زمان را بررسی کرد.
ممکن است مقدار هدف تعیین شده به شکل میانگین/میانهی همهی روشها یا با یک روش مرجع برای نمونههای تبادلناپذیر رضایتبخش به نظر آید زیرا بر حسب خوشاقبالی نتایج گروههای مختلف، معیارهای پذیرفته بودن را برآورده میکند. با وجود این، هیچ قاعدهی علمی مستحکمی در پس چنین رویکردی نیست. اگر نتایج یک روش در مقایسه با هدف حاصل از میانگین/میانه یا روش مرجع پذیرفته نشود یک توضیح بجا این خواهد بود که احتمال دارد اندازهی اختلاف وابسته به زمینه برای آن روش بیش از دیگر گروهها بوده است. بنا بر این، ناکامی یک روش در همخوانی با هدفهای حاصل از هر یک از این دو شکل، دلیل قطعی بر آن نیست که نتایج حاصل از سنجش نمونههای بالینی نیز غیرقابل قبول هستند. با وجود این، اختلاف آشکار ممکن است ناشی از اختلاف وابسته به زمینه نباشد، و برای مشخص کردن این که آیا چنین اختلافی در سنجش نمونههای بیماران نیز دیده میشود باید بررسی بیشتری انجام داد.
معیارهای پذیرش نتایج EQA
پیرامون هر مقدار هدف، مرزها یا معیارهای کیفیت تعیین میشود تا بتوان عملکرد شرکتکنندگان را بررسی کرد. به طور کلی مرزهای برنامههای EQA به سه شکل قانونی، آماری، یا بالینی تعیین میشوند.
محدودههای قانونی مانند CLIA88 در امریکا، [11]RiliBÄK در آلمان و [12]SPM در اسپانیا محدودههای بازتری هستند و هدف از برقراری آنها شناسایی عملکردهایی است که به اندازهای ضعیف هستند که باید متوقف شوند.
مرزهای آماری (مثلا 2 یا 3 انحراف معیار پیرامون میانگین) بر این فرض نانوشته بنا شدهاند که روشهای اندازهگیری موجود، از نظر برآورده کردن کیفیت مورد نیاز در بالین مناسب هستند و کافی است عملکرد یک آزمایشگاه با دیگران همخوانی داشته باشد. معیارهای آمار-بنیان این کاستی را دارند که ممکن است مرزهای پذیرش برای همهی گروهها یکسان نباشد. مرزهای پذیرش برای گروههای دارای روشهای غیردقیق وسیع خواهد بود و انگیزهی شرکتکنندگان آن گروه برای انتقال به روشهای بهتر کم خواهد بود. این در حالی است که ممکن است این محدودههای آماری بسیار بازتر از نیاز بالین باشند و در نتیجه عملکرد بسیاری از شرکتکنندگان این گروهها در حالی پذیرفته میشود که از نظر نیاز بالین عملکرد نامناسبی دارند. از سوی دیگر، یک گروه دارای روشی بسیار دقیق محدودهی پذیرش بسیار بستهای خواهد داشت که ممکن است باریکتر از نیازهای بالینی باشد؛ و این یعنی برخی از شرکتکنندگان آن گروه در برآورده کردن چنان معیارهای سختگیرانهای ناکام خواهند بود در حالی که عملکرد ایشان برای مراقبت بالینی مناسب است.
معیارهای بالین-بنیان، مثلا محدودههای تعیین شده بر اساس اختلافی که ممکن است بر تصمیمهای بالینی تاثیرگذار باشد یا محدودههای بنا شده بر نوسان زیستی، بهترین شکل هستند چون کیفیت مورد نیاز بالین را مد نظر قرار میدهند؛ اما تعیین چنین محدودههایی مشکل است. به عنوان نمونهای از مرزهای بالینی میتوان به معیار پذیرش نتایج HbA1c در برنامهی EQA به وسیلهی CAP[13] اشاره کرد. در این برنامه، برای فراهم کردن کیفیت مورد نیاز بالین برای استفاده از نتایج A1c در تشخیص دیابت، خطای کل مجاز برای سنجش این آنالیت برابر 6% در تعیین شده است. مرزهای بالینی مبتنی بر نوسان زیستی در حال حاضر در کشورهای هلند و استرالیا استفاده میشود. در دیگر کشورها از جمله آلمان نیز استفاده از این مرزها مدنظر قرار گرفته است. در طبقهبندی کنفرانس استکهلم 1999 برای معیارهای کیفیت در آزمایشگاه پزشکی، مرزهایی که بر اساس تاثیر عملکرد در هر وضعیت خاص بالینی تعیین شدهاند در برترین جایگاه هستند و مرزهای بالینی مبتنی بر نوسان زیستی در جایگاه دوم قرار دارند، در حالی که مرزهای آماری در پایینترین سطح این طبقهبندی یعنی جایگاه پنجم قرار دارند.
در سنجش نمونههای EQA منابعی برای نوسان نتایج وجود دارد که فقط بر نتایج EQA تاثیرگذار هستند و بر نتایج بیماران تاثیر ندارند و این سبب میشود که مرزهای پذیرش بازتر از آن مقداری باشد که بر اساس نیازهای بالینی لازم است. به عنوان مثال، تاثیر احتمالی ناشی از تخریب جزئی طی انتقال و نگهداری مواد کنترل متفاوت است با آنچه که برای جمعآوری و نگهداری نمونههای بیماران در شرایط بالینی رخ میدهد. در مورد مواد تبادلناپذیر، اندازهی اختلاف وابسته به زمینه ممکن است برای شماره گروههای مختلف یک معرف متفاوت باشد و بنا بر این پراکندگی دیده شده در نتایج کنترل بیش از آن مقداری باشد که در سنجش نمونههای بالینی دیده میشود. وجود این منابع نوسان از اشکالاتی است که باید سازماندهندگان برنامههای EQA تلاش کنند تا حد ممکن از آنها بکاهند تا بتوان مرزهای پذیرش را بستهتر در نظر گرفت و اختلاف نتایج را هرچه بیشتر به حساب عملکرد آزمایشگاه گذاشت و از این طریق بر توانایی خطایابی برنامه افزود.
محدودههای پذیرش باید به عنوان مرزهای مجاز برای “خطای کل” در نظر گرفته شوند زیرا هر سه عامل عدمصحت، عدمدقت و عدماختصاصیت یک روش میتواند در اختلاف یک نتیجهی منفرد با هدف نقش داشته باشند. چنانچه برنامهی EQA طوری طراحی شده باشد که نمونههای کنترل به تکرار سنجیده شوند ممکن است تعیین مرزهای پذیرش جداگانه برای عدم صحت و عدم دقت مناسب باشد. همچنین مهم است در نظر داشت که در بیشتر برنامهها، مرزهای پذیرش معیار حداقلی برای شناسایی عملکرد ضعیف هستند. بنا بر این، برآورده شدن چنین معیارهایی نه نشانهی عملکرد مطلوب آزمایشگاه است و نه دلیلی است بر تامین کیفیت مورد نیاز بالین. ممکن است برای بررسی این که آیا نتایج به دست آمده نیازهای بالینی را برآورده میسازند یا نه، مرزهای جداگانهای لازم باشد.
استفاده از نتایج EQA برای ارزیابی عملکرد آزمایشگاه
اندازهگیری و گزارش نتایج
به طور کلی برنامههای EQA از شرکتکنندگان میخواهند که نمونههای EQA را طوری که گویی نمونهی بیمار است آزمایش کنند. هیچ تلاشی نباید صورت بگیرد برای به دست آوردن “بهترین” نتایج از طریق سنجش چندباره یا سنجش بلافاصله پس از پایش کیفیت داخلی یا کالیبراسیون. چنین کارهایی به هدف اصلی برنامههای EQA که ارزیابی عملکرد آزمایشگاه در ارتباط با نمونههای معمول بیماران است آسیب میرساند.
در مراکزی که نمونههای بیماران با بیش از یک روش/دستگاه آزمایش میشود ممکن است آزمایشگاه به منظور یکسانسازی نتایج حاصل از دستگاههای گوناگون، کالیبراسیون را از وضعیتی که سازندهی تعیین کرده است تغییر داده و بر اساس روش/دستگاه دیگری تنظیم کرده باشد. هنگام سنجش نمونههای EQA با روش/دستگاهی که کالیبراسیون آن تعدیل شده است، چنانچه نمونهی کنترل تبادلپذیر است باید نتایج به دست آمده را در همان وضعیت کالیبراسیون تعدیلشده گزارش کرد زیرا تغییر کالیبراسیون به منظور اصلاح نتایج بیماران صورت گرفته است و با استفاده از کنترلهای تبادلپذیر میتوان درستی این تغییر را بررسی کرد. اما چنانچه مادهی کنترل تبادلناپذیر است باید نتایج را ابتدا به وضعیت کالیبراسیون پیشنهادی سازنده برگرداند و سپس گزارش کرد تا هنگام بررسی آنها با گروه مربوط اشکال ایجاد نشود.
پردازش دادهها و گزارش عملکرد
پردازش مناسب و قابل اعتماد دادهها یکی از الزامات اساسی هر برنامهی EQA است. این کار دارای جنبههای عملی و نظری است که باید در نظر گرفته شود. کاهش دادهها به شاخصهای آماری باید با روشهایی که مناسب برنامه است انجام شود. چنانچه در برنامهای از هدف توافقی استفاده میشود، باید ارزش نسبی میانگینها و میانهها بررسی شود و برای حذف دادههای پرت از روش آماری مستحکم و کارآمدی مانند روش Hely استفاده شود. سامانهای که برای امتیازدهی انتخاب میشود باید مستحکم و از نظر بالینی مربوط باشد و تحت تاثیر عملکرد دیگر شرکتکنندگان قرار نگیرد. یک سامانهی خوب این امکان را فراهم میکند که هم عملکرد هر شرکتکنندهی منفرد و هم عملکرد کلی همهی شرکتکنندگان را در طول زمان و در گسترهی جغرافیایی بررسی کرد.
تفسیر نتایج EQA در مورد نمونههای تبادلپذیر (ارزیابی درستی-بنیان)
نمونههای تبادلپذیر این حسن را دارند که رابطهی بین نتایج آنها در روشهای گوناگون، نظیر رابطهای است که بین نتایج بیماران وجود دارد. در نتیجه، آزمایشگاه میتواند از مقایسهی نتیجهاش در EQA با هدفی که به وسیلهی روش مرجع یا روش مقایسهای منتخب تعیین شده است درستی نتایج بیماران را مستقیما بررسی کند. امروزه چنین سازوکاری ارزیابی درستی-بنیان نامیده میشود.
نمونههای تبادلپذیر همچنین این امکان را فراهم میسازند که آزمایشگاه بتواند همخوانی نتیجهاش با دیگر روشها و همچنین پراکندگی نتایج درون یک گروه روشی و بین همهی روشها را که بازتاب شرایط نمونههای بیماران است بررسی کند.
در صورت توزیع تکراری نمونهها میتوان برآورد قابل اعتمادی از عدمدقت آزمایشگاه در طول زمان به دست آورد.
تفسیر نتایج EQA در مورد نمونههای با تبادلپذیری نامشخص
در این مورد به دلیل محدودیتهای ناشی از تبادلناپذیری، نتایج هر آزمایشگاه با میانگین/میانهی یک گروه از روشها که انتظار میرود اختلاف وابسته به زمینهی یکسان یا خیلی نزدیک به هم داشته باشند مقایسه میشود. ارزیابی گروهی این اجازه را نمیدهد که بتوان درستی نتیجه را به طور مستقیم در مقابل روش مرجع، روش مقایسهای منتخب، یا میانگین/میانهی همهی شرکتکنندگان (یا همهی روشها) ارزشیابی کرد. با وجود این، ارزیابی گروهی این امکان را برای آزمایشگاه فراهم میکند که بتواند بررسی کند آیا یک روش را مطابق با ویژگیهای سازنده و هماهنگ با بقیهی آزمایشگاههای بهرهمند از آن فناوری به کار میبندد یا نه. در این حالت، تضمین ردیابیپذیری آن روش به بالاترین سطح سامانهی اندازهگیری به عهدهی سازنده است. چنانچه بتوان اطمینان داشت که سازندهی یک روش آن را به درستی کالیبر کرده است و آن روش قابل ردیابی به ماده و/یا روش مرجع است، در این صورت ارزشیابی تطابق عملکرد آزمایشگاه با ویژگیهای سازنده، به طور غیرمستقیم ارزشیابی درستی نتایج بیماران است. با وجود این، باید در نظر داشت که چنانچه همهی کالیبراتورهای توزیع شده در یک منطقه مشکل داشته باشند، یک برنامهی EQA تبادلناپذیر ممکن است نتواند اشکال کالیبراسیون را شناسایی کند زیرا در این صورت اگرچه یک آزمایشگاه ممکن است سازگاری خوبی با گروه داشته باشد اما اشکال در این جاست که آن آزمایشگاه همراه با بقیهی اعضای آن گروه در اشتباه است و کاری هم از مواد تبادلناپذیر برای شناسایی چنین اشکالی برنمیآید. به طور کلی، نمونههای تبادلناپذیر برآورد خوشبینانهای از عملکرد ارائه میکنند.
در صورت توزیع تکراری نمونهها، همانند نمونههای تبادلپذیر، میتوان برآورد قابل اعتمادی از عدمدقت آزمایشگاه در طول زمان به دست آورد.
استفاده از EQA برای ارزیابی عملکرد روشها
EQA با استفاده از نمونههای تبادلپذیر (ارزیابی درستی-بنیان)
برنامههای EQA که از نمونههای تبادلپذیر استفاده میکنند برای سازندگان روشهای تشخیصی، برای آزمایشگاههایی که روشهای سنجش را خودشان تهیه میکنند و برای فعالیتهای عیارمندسازی/هماهنگسازی اهمیت ویژهای دارند.
نتایج برنامههای تبادلپذیر رابطهای را که از نتایج بیماران انتظار میرود بازتاب میدهند زیرا هیچ اختلاف وابسته به زمینهی چشمگیری وجود ندارد. به منظور ارزیابی یکدستی نتایج بیماران در سنجش با روشهای گوناگون، میتوان مقادیر میانگین/میانهی روشهای گوناگون را با یکدیگر و همچنین با نتایج حاصل از روش مرجع، روش مقایسهای منتخب، یا میانگین/میانهی همهی شرکتکنندگان مقایسه کرد. در نتیجهی چنین مقایسههایی، روشهایی که نتایج خیلی متفاوت تولید میکنند شناخته میشوند و شرکتهای سازنده میتوانند کالیبراسیون آنها را اصلاح کنند. در برنامههای بهرهمند از مواد تبادلپذیر، انحراف معیار روشها تحت تاثیر همان عواملی قرار خواهند گرفت که بر عدمدقت نمونههای بیماران تاثیرگذار هستند و بنا بر این، انحراف معیار گروه شاخصی است برای ارزیابی یکدستی بینآزمایشگاهی در نتایج بیماران. به بیان کوتاه، نتایج چنین برنامههایی بهترین اطلاعات را در بارهی کارآمدی سامانهی پایش کیفیت یک سازنده در انتقال ردیابیپذیری کالیبراسیون و نیز تامین یکدستی نتایج بین کاربران مختلف فراهم میکند و کاربران را از این موضوع که کدام فناوریها صحت بهتر و یکدستی بینآزمایشگاهی بهتری دارند آگاه میکند.
با استفاده از دادههای EQA تبادلپذیر میتوان به تشکلهای حرفهای بالینی نسبت به تصمیمهایشان در بارهی استفاده از نتایج آزمایشگاهی اطلاعرسانی کرد. به عنوان مثالهایی اخیر از آزمایشهایی که در امریکا و اروپا کیفیت آنها با استفاده از برنامههای تبادلپذیر بررسی شده، بهبود داده شده و سپس بازبررسی شده است میتوان از کراتینین سرم برای محاسبهی سرعت گلومرولی برآوردی (eGFR) و هموگلوبین A1c برای تشخیص و پیگیری دیابت نام برد. ارائه چنین گزارشهایی از سوی سازمانهای EQA به جامعهی پزشکی میتواند بسیار سودمند باشد زیرا به کاربران نتایج آزمایشگاهی نشان میدهد که تا چه اندازه میتوانند به این نتایج اطمینان کنند و نیز تا چه اندازه میتوانند نتایج آزمایشهای یک بیمار را که در زمانها و/یا محلهای گوناگون انجام شده است با یکدیگر مقایسه و بر اساس آن در بارهی روند تغییرات بیماری اطلاع کسب کنند.
EQA با استفاده از نمونههای با تبادلپذیری نامشخص
نظر به اینکه بنا بر گزارشها در تقریبا 50% از چنین موادی اختلاف وابسته به زمینه دیده شده است و نیز نامشخص بودن اندازهی چنین اختلافهایی، نمیتوان از نتایج چنین برنامههایی برای تعیین رابطهی عددی بین میانگینهای گروهی حاصل از روشهای مختلف و نیز رابطهی میانگینهای گروهی با یک روش مرجع، استفاده کرد. در جدول 1 مثالی از نتیجهگیری اشتباه بر اساس یک برنامهی EQA تبادلناپذیر برای ویتامین D ارائه شده است. اختلاف آشکاری که در این مثال بین نتایج گروههای مختلف در سنجش یک نمونهی مرسوم تبادلناپذیر دیده میشود نتیجهی تفاوت در اندازهی اختلاف وابسته به زمینه برای روشهای مختلف است، زیرا همهی این گروهها در سنجش یک نمونهی تبادلپذیر تقریبا نتایج یکسانی به دست دادهاند. در مواردی که اختلاف وابسته به زمینه برای روش خاصی معین شده باشد، ممکن است بتوان از یک فاکتور اصلاح برای حذف بخشی از اختلاف که وابسته به زمینه است استفاده وسپس عملکرد آن روش را بررسی کرد.
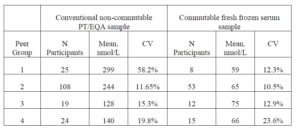
جدول 1- مقایسهی سنجش 25OH Vitamin D با نمونههای EQA تبادلپذیر و تبادلناپذیر.
بررسی CAP-2009Y-A
بررسی نمونههای کنترل با روش MS تایید کرد که هر دو نمونه دارای 100% ویتامین D3 هستند و بنا بر این نمیتوان اختلاف بین نتایج گروهها را به اختلاف در حساسیت روشها برای D2 و D3 نسبت داد.
اگرچه ارزیابی گروهی میتواند ابزار خوبی برای بررسی همخوانی نتایج یک گروه باشند، اما این همخوانی نمیتواند تضمینی بر درستی نتایج باشد؛ یعنی ممکن است در حالی که گروهها نتایج بسیار یکدستی دارند در همان حال همهی نتایج یک یا چند گروه نادرست باشد. نمونهای از این وضعیت در رابطه با سنجش متانفرینهای ادراری در امریکا، در نوشتهای با عنوان “به دقت اشتباه؟” در مجلهی کلینکال کمیستری ارائه شده است. برنامهی EQA متانفرینهای در امریکا برنامهای تبادلناپذیر است و بنا بر این نتایج شرکتکنندگان با میانگین گروه مقایسه میشود. در سه دور پی در پی در 2004/2003 نتایج آزمایشگاه غدد مایوکلنیک غیرقابل قبول و بیش از دیگر شرکتکنندگان بود. تا پیش از سال 2003 آزمایشگاه مایو هم مانند دیگران از کالیبراتور تجاری ساخت شرکت Bio-Rad استفاده میکرد، اما در آن سال با تهیهی تهیهی متانفرین و نورمتانفرین از شرکت سیگما، خودشان کالیبراتور تهیه کرده بودند. بررسیهای گستردهی بخش پایش کیفیت آزمایشگاه مایو نشان داد که نتایج ایشان درست است و اشکال در کالیبراتور Bio-Rad است. در واقع بسیاری از آزمایشگاههایی که طی سالها از این کالیبراتور استفاده میکردند نتایجی به دست میآوردند که به رغم یکدستی گروهی بسیار خوب، از مقدار درست فاصله داشت.
طبقهبندی برنامههای EQA
امروزه برنامههای EQA را بر اساس کیفیت توانایی آنها در ارزیابی عملکرد به شش سطح تقسیم میکنند. توانمندی یک برنامه به سه ویژگی بستگی دارد: تبادلپذیری نمونه، فرآیند ارزشگذاری، و فرستادن نمونههای تکراری (جدول 2).
برنامههای سطح 1 بهترین هستند زیرا در چنین برنامههایی از نمونههای تبادلپذیر استفاده میشود، مقادیر هدف با استفاده از روشهای مرجع تعیین میشود و نمونهها به صورت تکراری فرستاده میشوند. این برنامهها میتوانند روشها را از نظر تکرارپذیری، ردیابیپذیری کالیبراسیون و یکدستی نتایج بین آزمایشگاهها و بین روشها مورد بررسی قرار دهند. در واقع این برنامهها با داشتن توانایی ارزیابی ردیابیپذیری کالیبراسیون این امکان را فراهم میکنند که بتوان “عیارمندی” سنجش آنالیتها را بررسی کرد. همچنین به دلیل فرستادن نمونههای تکراری این قابلیت را دارند که تکرارپذیری درونآزمایشگاهی را بررسی کنند.
برنامههای سطح 2 همان خصوصیات سطح 1 را دارند با این تفاوت که چون نمونههای تکراری نمیفرستند بنا بر این نمیتوانند تکرارپذیری درونآزمایشگاهی را بررسی کنند.
برنامههای سطوح 3 و 4 نیز از نمونههای تبادلپذیر استفاده میکنند اما چون از روشهای مرجع برای تعیین مقادیر هدف استفاده نمیکنند توانایی این برنامهها برای ارزیابی روشها محدود است به بررسی یکدستی نتایج، یعنی ارزیابی “هماهنگی” سنجش آنالیتها؛ که البته توانایی ارزشمندی است.
برنامههای سطوح 5 و 6 از نمونههایی که احتمال دارد تبادلناپذیر باشند استفاده میکنند و ناگزیر توان ارزیابی آنها محدود است به فقط مقایسهی گروهی و نمیتوانند در بارهی اختلاف بین روشهای گوناگون اطلاعاتی به دست دهند.

جدول 2- طبقهبندی توانمندی برنامههای EQA
بی گمان همهی برنامههای EQA باید سطح 1 باشند. با وجود این، اجرای چنین برنامههایی با چالشهایی روبرو است ناشی از:
- جنبههای فنی مانند نبود روشهای مرجع، نبود مواد مرجع گواهینامهدار، ناتوانی در تولید مواد تبادلپذیر؛
- ملاحظات عملی مانند سختی تولید نمونههایی که همهی بازهی اندازهگیری را در بر بگیرند، پیچیدهگی تدارکات تهیه و توزیع نمونههای تازهی یخزده؛
- محدودیتهای روانشناختی مانند ناآگاهی از عواملی که در کیفیت برنامههای EQA اهمیت دارند و یا عدم تمایل به پذیرش آنها؛
- مسائل اقتصادی زیرا تعیین مقادیر هدف با روشهای مرجع و توزیع نمونههای تبادلپذیر فرآیندی پرهزینه است.
نظر به ارزشمندی برنامههای سطح 1 و 2، تلاشهای موفقیتآمیزی برای غلبه بر چالشهای پیش روی اجرای چنین برنامههایی انجام شده است و از سالها پیش استفاده از چنین برنامههایی در برخی کشورها آغاز شده است. در زیر برنامهی EQA در هلند، بریتانیا و امریکا که در آنها سطوح مختلف EQA انجام میشود به طور مختصر بیان میشود.
برنامهی EQA در هلند
نظر به اهمیت روزافزون همسانی نتایج آزمایشگاهها، از سالهای پایانی دههی 1990 در هلند تلاشهای گستردهای برای عیارمندسازی/هماهنگسازی نتایج بیوشیمی با عنوان “کالیبراسیون 2000” آغاز شد. دستاندرکاران این فعالیت برای این که بتوانند اجرای همسانسازی و سپس برقرار ماندن آن را به طور پیوسته پایش کنند خود را ناگزیر از اجرای یک برنامهی EQA سطح بالا دیدند. بنابراین، به دنبال تلاشی گسترده و چندساله برای طراحی، ارزشیابی و اجرای یک برنامهی مناسب، از سال 2005 به این سو در برنامهی EQA هلند (SKML[14]) از یک برنامهی سطح 1 برای ارزیابی آنالیتهای بیوشیمی استفاده میشود.
در برنامهی SKML طی یک دورهی یک ساله برای هر آنالیت 12 جفت نمونهی تبادلپذیر به آزمایشگاهها فرستاده میشود (روی هم 24 نمونه). هر جفت نمونه از تقسیم یک نمونهی واحد به دو بخش تهیه شده است که یک بخش آن برای سنجش در نیمهی اول سال و بخش دیگر برای نیمهی دوم سال در نظر گرفته شده است. آزمایشگاه نمونههای دریافتی را با تناوب یک نمونه در هر دو هفته آزمایش میکند.
برای تهیهی نمونههای تبادلپذیر از دستورکاری که بسیار به دقت و طی فرآیندی هزینهبر و زمانبر تهیه و ارزشیابی شده است استفاده میشود. به طور کوتاه، برای تهیهی نمونهها ابتدا دو انباشتهی سرمی با غلظت طبیعی از باقیماندههای نمونههای بیماران تهیه میشود. به یکی از از این انباشتهها نمونههای غیرطبیعی، مواد معدنی، آنزیمهای انسانی ریکامبیننت و آلبومین انسانی افزوده میشود تا یک انباشتهی با غلظت غیرطبیعی بالا به دست آید. سپس از ترکیب این دو انباشتهی اصلی به نسبتهای مختلف، 10 غلظت بینابینی تهیه میشود تا روی هم 12 سطح برای هر آنالیت در دسترس باشد طوری که سراسر بازهی بالینی پوشش داده شود. نمونهها پس از تهیه در دمای 84o C- نگهداری و روی یخ خشک به آزمایشگاهها فرستاده میشود و در آزمایشگاه نیز تا زمان سنجش در دمای < 70o C- نگهداری میشود.
برای ارزشگذاری نمونهها، مقادیر هدف چربیها شامل کلسترول، تریگلیسریدها، HDL و LDL در آزمایشگاه CDC (آزمایشگاههای شبکهای NCEP) و مقادیر هدف آنالیتهای دیگر در آزمایشگاههای فهرست JCTML تعیین میشود. چون برای سنجش آلبومین، ALP، فسفات و اوره در حال حاضر روش مرجع در دسترس نیست، مقادیر هدف این آنالیتها به صورت توافقی (میانگین همهی شرکتکنندگان) تعیین میشود (جدول 3). اگرچه همانطور که گفته شد تبادلپذیری این مواد پیش از شروع برنامه و در فرآیندی چند ساله و بر اساس دستورکار CLSI-EP30A ارزشیابی شده است، به رغم این، هر ساله یک نمونه نیز بر اساس دستور کار CLSI-C37A تهیه و همراه نمونهها به آزمایشگاهها فرستاده میشود تا تبادلپذیر بودن مواد تهیه شده به طور مستمر پایش شود.

جدول 3 – فرآیند ارزشگذاری آنالیتهای بیوشیمی (غیر از لیپیدها) در SKML
در این برنامه آنالیتهای غیر لیپیدی به دو گروه تقسیم میشوند: آنالیتهایی که برای آنها از روشهای مرجع برای ارزشگذاری استفاده میشود (13 آنالیت که خود به زیرگروههای مواد معدنی، سوبستراها و آنزیمها تقسیم میشوند) و آنالیتهایی که برای آنها روشهای مرجع در دسترس نیست (4 آنالیت). روش ارزشگذاری آنالیتهایی که در این جدول نیامده عبارتند از: آلفا-آمیلاز (روش مرجع IFCC، آزمایشگاه مرجع بیمارستان Haga)، بیلیروبین (روش مرجع IFCC، آزمایشگاه مرجع DGKL هانوور)، پروتئین (روش مرجع بیوره، آزمایشگاه مرجع INSTANS دوسلدورف)، آهن (بدون روش مرجع، قابل ردیابی به مادهی مرجع NIST-SRM 937) و لیپاز (هدف توافقی).
این برنامه با داشتن 12 سنجش دوتایی برای هر آنالیت که سراسر بازهی فیزیولوژیک و پاتولوژیک را پوشش میدهد این اجازه را میدهد که بتوان ویژگیهای دوگانگی، خطیبودن و بازیافت را بررسی کرد. از دیگر نقاط قوت این برنامه، محدودههای مجاز به کارگرفته شده در آن است. در این برنامه از محدودههای بالینی که بر مبنای نوسان زیستی تعیین شده است به عنوان محدودهی پذیرش عملکرد استفاده میشود. در پایان هر دوره، نتایج آزمایشگاه در مقابل مقادیر هدف بر روی یک نمودار اختلاف نمایش داده میشود (شکل 3). در نمودار اختلاف، بر روی محور عرضها اختلاف نتیجهی آزمایشگاه با هدف و بر روی محور طولها غلظت هدف نمایش داده میشود. بر اساس خطای کل مجاز مطلوب (Desirable TEa) حاصل از دادههای نوسان زیستی، ناحیهی پذیرش پیرامون خط عدم اختلاف (y = 0) تعیین میشود (ناحیهی سبز رنگ در شکل 3). چون محدودهی پذیرش این برنامه یک محدودهی بالینی است بنا بر این محل قرار گرفتن نتایج آزمایشگاهها و روشها نسبت به این محدوده نمایانگر عملکرد آنها در تامین کیفیت مورد نیاز بالین است. برای تعیین عدم صحت هر آنالیت، میانگین 24 نتیجهی به دستآمده در طول سال با میانگین مقادیر هدف مقایسه میشود و عدمصحت آزمایشگاه با استفاده از اختلاف بین این دو میانگین محاسبه میشود .
برای بررسی عدمدقت آزمایشگاه، نتایج آزمایشگاه در مقابل میانگینهای گروه روی نمودار برده میشود و سپس خط معادلهی برگشت خطی (linear regression) رسم شده و انحراف معیار پراکندگی نتایج آزمایشگاه پیرامون این خط، یعنی انحراف معیار باقیماندهها (SDxy)، محاسبه میشود (در شکل 3 خط برگشت به صورت خط سیاهرنگی که از بین نتایج میگذرد دیده میشود). از آنجائی که این نمونهها در روزهای مختلف و در یک دورهی یک ساله سنجیده شدهاند انحراف معیار محاسبه شده نمایانگر نوسان درونآزمایشگاهی (SDWL) بلندمدت است.
با استفاده از عدمصحت محاسبه شده و SDWL در سطح غلظت میانگین، خطای کل آزمایشگاه محاسبه و با TEa مقایسه میشود تا عملکرد یک سالهی آزمایشگاه مشخص شود.
بررسی ارزشمند دیگری که انجام میشود ارزیابی عملکرد کلی روشهای سنجش بر اساس کیفیت مورد نیاز بالین است. برای این کار، محدودهای که 90% نتایج حاصل از همهی روشها در آن قرار میگیرد مشخص میشود (محدودهی آبی رنگ در شکل 3). این محدوده نمایانگر وضعیت عملی فناوریهای موجود است. همانطور که در شکل زیر دیده میشود برای CK این محدوده از محدودهی پذیرش بالینی بستهتر است که نشاندهندهی آن است که روشهای مورد استفاده به خوبی میتوانند کیفیت مورد نیاز بالین را تامین کنند. برعکس، در مورد کلسیم محدودهی فناوری از محدودهی بالینی بازتر است و این یعنی روشهای مورد استفاده در بسیاری از موارد نمیتوانند کیفیت بالینی لازم را تامین کنند.
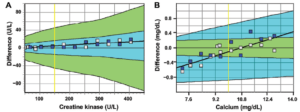
شکل 3 – مثالی از گزارش سالانهی نتایج کراتینین کیناز CK) و کلسیم (Ca) در برنامهی SKML
ناحیهی سبز نمایانگر خطای کل مجاز بالینی است. ناحیهی آبی محدودهای است که 90% نتایج همهی آزمایشگاهها در آن قرار دارند و نمایانگر وضعیت کنونی فناوری سنجش است. روی محور X مقادیر هدف نمونهها که با روش مرجع تعیین شده است قرار دارد و روی محور Y اختلاف نتیجهی آزمایشگاه با مقدار هدف قرار دارد. خط زرد عمودی نمایانگر سطح تصمیمگیری بالینی است. مربعهای سفید نتایج نیمهی اول سال هستند و مربعهای آبی نمایانگر نتایج نیمهی دوم سال هستند.
در سنجش CK نتایج این آزمایشگاه برای هر 24 نمونه (12 سطح) درون محدودهی مجاز بالینی قرار میگیرد و قابل قبول است. در مورد سنجش کلسیم چنین نیست و تعدادی از نتایج بیرون از ناحیهی سبز قرار میگیرد. نتایج ناپذیرفتهی زیر mg/dL 9 اختلاف منفی دارند و برعکس نتایج ناپذیرفتهی بالاتر از 11 اختلاف مثبت دارند؛ این وضعیت نتیجه وجود خطای سامانمند نسبی برای سنجش کلسیم در این آزمایشگاه است که به خوبی از شیب صعودی خط گرایش پیداست.
از مقایسهی وضعیت ناحیهی آبی نسبت به ناحیهی سبز دیده میشود که در مورد CK محدودههای مبتنی بر فراوانی توزیع از محدودههای پذیرش بالینی بستهتر هستند؛ یعنی عملکرد روشهای موجود میتواند کیفیت مورد نیاز بالین را برآورده کند. در مورد کلسیم وضعیت برعکس است و محدودههای مبتنی بر توزیع از محدودههای پذیرش بالینی بازتر هستند؛ یعنی روشهای موجود نمیتوانند کیفیت بالینی مورد نیاز را تامین کنند. فناوریها کنونی برای سنجش کلسیم علاوه بر پراکندگی بینآزمایشگاهی زیاد، همانطور که از شیب صعودی ناحیهی آبی پیداست از خطای سامانمند نسبی نیز رنج میبرند. نکتهی قابل توجه در شکل B این است که آن تعداد از نتایج کلسیم این آزمایشگاه که بیرون از محدودهی بالینی هستند همچنان درون محدودهی 90% روشها هستند. اگر بنا بود به جای استفاده از مقادیر حاصل از روش مرجع و مرزهای بالینی، از میانگین گروهی و مرزهای آماری برای ارزیابی وضعیت این آزمایشگاه استفاده شود عملکرد وی برای کلسیم کاملا پذیرفته میشد و کاستی عملکرد آن از نظر تامین کیفیت مورد نیاز بالین از نظر دور میماند.
SKML علاوه بر اجرای برنامهی ارزیابی کیفیت خارجی، در قالب برنامهی کالیبراسیون 2000 تلاش گستردهای را برای ردیابکردن نتایج سنجش آنزیمها در آزمایشگاههای هلند به روشهای مرجع IFCC انجام داده است. در این راستا با همکاری IFCC نمونهی “هماهنگساز” آنزیمی تهیه و بین آزمایشگاهها توزیع کرده است تا آزمایشگاهها روشهای خود را با استفاده از آن تنظیم کنند. مقایسهی نتایج سال 2005 (پیش از توزیع هماهنگساز) و سال 2010 نشان میدهد که این اقدام بسیار ثمربخش بوده است.
توانمندی برنامهی SKML به حدی است که در سال 2011 در قالب پژوهشی برای ارزیابی وضعیت عیارمندی/هماهنگی نتایج آزمایشگاهی چند آنالیت در سطح اروپا، سازماندهندگان آن پژوهش از برنامهی SKML استفاده کرده و 6 نمونهی بیوشیمی این برنامه را همزمان با توزیع در هلند، در آزمایشگاههایی از کشورهای انگلستان، اسپانیا و پرتغال توزیع کردند. نتایج این بررسی نشان داد که آزمایشگاههای هلند در مقایسه با سه کشور دیگر بهترین وضعیت را از نظر دقت قابلقبول، صحت قابلقبول و خطای کل قابلقبول دارند و مجریان آن را به این نتیجه رساند که “اجرای برنامهی ارزیابی کیفیت خارجی با استفاده از نمونههای تبادلپذیر که با روشهای مرجع تعیین هدف شدهاند نقشی اساسی در پایش عملکرد آزمایشگاههای بالینی و در نتیجه، شناسایی درست خطراتی که متوجه بیمار است دارند؛ و این دلیل دیگری است برای این که برنامههای پایش کیفیت کنونی را به سوی برنامههای سطح 1 ارتقا دهیم”. پس از اجرای این برنامه، مجریان ارزیابی کیفیت خارجی اسپانیا تصمیم گرفتند که هر ساله آزمایشگاههای اسپانیا را در سنجش تعدادی از نمونههای SKML شرکت دهند (برنامهی پایش کیفیت اسپانیا با توزیع تکراری نمونههای تبادلناپذیر اجرا میشود و بنابراین یک برنامهی سطح 5 است).
برنامهی EQA در بریتانیا
برنامهی ارزیابی کیفیت خارجی انگلستان (UK NEQAS[15]) از سال 1969 آغاز به کار کرده است و طراحی آن بازتابدهندهی طرح و هدف WHO برای برنامههای ارزیابی کیفیت خارجی است. هدف این برنامه علاوه بر ارزیابی عملکرد یک آزمایشگاه منفرد عبارت است از بررسی وضعیت کلی موجود (معیار عمومی عملکرد) و نیز عملکرد هر شیوهی سنجش (اصول روش، معرفها و دستگاهها).
برای معرفی این برنامه، فعالیتهای بخش بیوشیمی و هورمون این برنامه با عنوان “کیفیت بیرمنگام” (با نام پیشین آزمایشگاه EQA ولفسون) که عمدهترین بخش آن است به طور خلاصه معرفی میشود. کیفیت بیرمنگام همچنین “مرکز همکاری WHO برای تحقیق و خدمات مرجع در شیمی بالینی” است و در این نقش، وظیفهی هماهنگ کردن تحقیقات مشترک، ارتقاء ارزیابی کیفیت موثر در آزمایشگاههای بهداشتی و کمک به برپاداشتن برنامههای منطقهای و ملی EQA در سراسر جهان را به عهده دارد.
در دهههای اول شروع برنامه، بنا به دلایل عملی برای بیوشیمی نمونههای لیوفیلیزه، عموما با منشاء حیوانی، فرستاده میشد. برای آلبومین، هورمونها و پروتئینهای اختصاصی سرم، نمونههای مایع سرم انسانی (در صورت نیاز با نگهدارنده) فرستاده میشد. در حال حاضر در تهیهی نمونهها تاکید بر آن است که نمونههای انسانی با حداقل دستکاری و تغییر زمینه تهیه شود. برای کاستن از اثر زمینهای سعی میشود تعداد نمونههای اهداکنندگان در یک انباشته تا حد ممکن کم باشد، در صورت امکان فقط دارای آنالیتهای بومی باشد و هر نمونه برای پایش فقط یک یا چند آنالیت تهیه شود. به عنوان مثال، برای تهیهی نمونهی هورمونهای پپتیدی، سرم مایع از فیلتر 0.2 میکرون عبور داده میشود و به عنوان نگهدارنده کاتون 0.5% به آن افزوده میشود. استفاده از سدیم آزاید از چند سال پیش که روشن شد این ماده بر یکی از روشهای ایمونواسی برای سنجش LH و HCG تاثیر میگذارد متوقف شده است.
علاوه بر نمونههای معمول، به طور منظم انباشتههای خاصی تهیه میشود که بتواند مشکلات یا موارد علمی/بالینی خاص را بررسی کند. مثلا نظر به تفاوت روشهای ایمونولوژیک در شناسایی آنالیتهای دارای ساختار ناهمگن، در این برنامه برای جستجوی کامل و دقیق دلایل این اختلافهای وابسته به روش از نمونههایی که به دقت برای این منظور تهیه میشوند استفاده میشود. چنین نمونههایی میتوانند تاثیر احتمالی چنین اختلافهایی بر نتایج بیماران را نشان دهند. نمونههایی که فاقد آنالیت باشند یا غلظت بسیار کمی از آن را در برداشته باشند تهیه و برای بررسی حساسیت روشهای سنجش توزیع میشود. همچنین نمونههایی برای بررسی اختصاصیت با افزودن مداخلهگرهای فیزیولوژیک (مانند بیلیروبین) و غیرفیزیولوژیک (مانند داروها) و نمونههایی با غلظتهای بسیار بالا برای بررسی اثر هوک تهیه میشود.
نمونههای بیوشیمی با تناوب هر دو هفته یک بار توزیع میشود و آزمایشگاهها باید تا یک هفته پس از دریافت نمونه، جواب را برگردانند. نتیجهی ارزیابی نیز باید تا پیش ازتوزیع بعدی به آزمایشگاه گزارش شود. برای هورمونها به طور معمول ماهانه 3 تا 5 نمونه توزیع میشود. توزیع این تعداد نمونهی هورمونی، علاوه بر فراهم کردن امکان موشکافی عمیقتر جنبههای بالینی مربوط (مانند تداخل ناشی از ماکروپرولاکتین در روشهای سنجش پرولاکتین)، مبنای آماری محکمی برای ارزیابی انباشتی فراهم میکند. برای اطمینان از این که از منابع ناشناخته تغییرات اضافی به نتایج افزوده نمیشود، آزمایشگاه باید همهی نمونههای دریافت شده در یک توزیع را در یک روز، با یک شماره ساخت از معرف و در یک دور و با کالیبراسیون یکسان آزمایش کند. برای بیشتر برنامهها، فاصلهی زمانی بین بسته شدن یک توزیع و انتشار گزارش بررسی کمتر از 5 روز کاری است.
پیش از برقراری روشهای مرجع ، مقادیر هدف برای همهی آنالیتها به صورت توافقی (میانگین شرکتکنندگان) تعیین میشد. امروزه برای این که امکان ارزیابی و مقایسهی درستی-بنیان را فراهم باشد، برای آنالیتهایی که برای آنها روشهای مرجع وجود دارد (مانند IDMS برای بسیاری از آنالیتهای بیوشیمی عمومی و هورمونهای استروئیدی) مقادیر هدف با روشهای مرجع تعیین میشود. مقدار هدف نمونههای HbA1c به وسیلهی شبکهی آزمایشگاههای مرجع تعیین میشود. برای آنالیتهایی که روش/مادهی مرجع برای آنها وجود ندارد اما یک فرآوردهی مرجع پذیرفتهشدهی بینالمللی برای آنها وجود دارد (مثل TSH و HCG)، از این فرآوردهها در بررسیهای بازیافت/خطیبودن (رقیقسازی) برای ارزشیابی مقادیر هدف استفاده میشود. در مورد آنالیتهای برونزاد که به طور طبیعی در مایعات بدن وجود ندارند (مانند داروها)، مقادیر هدف با تعیین وزن مادهی افزوده شده محاسبه میشود. در فرآیند تعیین میانگین برای مقادیر هدف توافقی، از شیوهای که Hely در سال 1979 معرفی کرده است برای حذف دادههای پرت استفاده میشود. برتری این روش Hely بر شیوهی مبتنی بر انحرافمعیار در این است که روش Hely هم استحکام آماری بهتر و هم کارآمدی بیشتری دارد؛ یعنی هم دادههای پرت را در صورت وجود بهتر شناسایی و حذف میکند و هم دادههایی را که دادهی پرت واقعی نیستند به اشتباه حذف نمیکند. نکتهی قابل توجه در رابطه با مقادیر هدف توافقی این است که هرگز دستاندرکاران UK NEQAS فرض را بر این نگذاشتهاند که میانگین حاصل به طور خودکار “مقدار درست” است؛ بلکه از آغاز برنامه، برای اطمینان از قابل اعتمادبودن این مقادیر هدف، به طور پیوسته و منظم در طول سال با انجام مطالعات تکرارپذیری، بازیافت و بررسی مقدار پایه در نمونههای عاری از آنالیت و همچنین از طریق همکاری با دیگر برنامههای EQA، مقادیر هدف توافقی را ارزشیابی میکنند.
تا پیش از سال 1972، بررسی نتایج هر دور با استفاده از میانگین و نتایج همان دور و محاسبهی شاخص “انحراف معیار اختلافها” (SDD[16]) یا نمرهی Z (Z score) انجام میشد:
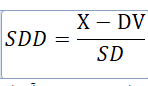
در این رابطه، X نشانهی نتیجهی آزمایشگاه، DV نشانهی ارزش منتصب[17] یا مقدار هدف و SD نشانهی انحراف معیار گروه است. شاخص SDD نشان میدهد که نتیجهی آزمایشگاه چند انحراف معیار و در چه جهتی از مقدار هدف دور است. مشکل این شیوه در آن بود که نمیشد عملکرد یک آزمایشگاه را مستقل از عملکرد دیگران بررسی کرد زیرا مقدار SD بستگی داشت به عملکرد آزمایشگاههای دیگر و همچنین به دلیل تغییر انحراف معیار در توزیعهای مختلف، ارزیابی تغییرات عملکرد در طول زمان ممکن نبود. برای رفع این کاستی، شیوهی امتیازدهی شاخص نوسان (VI[18]) ابداع و از سال 1972 به بعد ابتدا در UK NEQAS و به دنبال آن در برنامههای WHO مورد استفاده قرار گرفت.
محور اصلی سامانهی VI در استفاده از انحراف معیار ثابت در دورهای مختلف است. دستاندرکاران UK NEQAS بر اساس نتایج به دستآمده از توزیعهای مختلف در سال 1972، کوچکترین ضریب تغییرات برای هر آنالیت را به عنوان شاخص عملکرد قابل دستیابی با فناوریهای موجود انتخاب و “ضریب تغییرات منتخب” (CCV[19]) نامیدند. در سالهای بعد هم CCV آنالیتهای جدید بر همین اساس تعیین شده است. با فرض ثابت بودن CV در سراسر بازهی سنجش، عکس CCV در مقدار هدف ضرب میشود تا SD مناسب آن سطح به دست آید و سپس از آن برای محاسبهی فاصلهی نتیجهی آزمایشگاه از مقدار هدف استفاده میشود (رابطهی BIS در زیر). در سامانهی VI در هر دور برای هر آنالیت شاخصهای زیر حساب میشود:
- امتیاز شاخص اختلاف (BIS[20]):
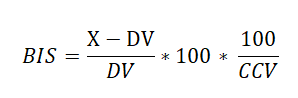
این شاخص، مشابه SDD، نشان میدهد که نتیجهی آزمایشگاه چند انحراف معیار و در چه جهتی از هدف دور است با این تفاوت که به جای استفاده از انحراف معیار گروهی از انحراف معیار محاسبه شده بر اساس CCV استفاده میشود، و تفاوت دیگر این که یک ضریب 100 در فرمول BIS گنجانده شده است تا اعداد اعشاری نداشته باشیم. ارقام بالاتر از 400 به صورت 400 گزارش میشود؛ بنا بر این بازهی BIS از 400- تا 400+ است.
- امتیاز شاخص نوسان (VIS[21]): این شاخص، قدر مطلق BIS است. هدف از محاسبهی VIS آن است که بتوانیم با استفاده از آن، خطای کل بلندمدت آزمایشگاه را حساب کنیم. بازهی VIS از صفر تا 400 است.
همانطور که پیشتر گفته شد هدف اصلی برنامههای EQA ارزیابی بلند مدت عملکرد است. در UK NEQAS با انباشت نتایج آخرین 10 توزیع (نتایج تقریبا تا 6 ماه پیش) برای هر آنالیت بیوشیمی شاخصهای زیر حساب میشود:
- امتیاز A؛ میانگین روندهی VIS (MRVIS[22]): این شاخص حاصل میانگین گرفتن از ده VIS آخر است و نمایانگر خطای کل آزمایشگاه در بلند مدت است؛ چون هم عدم صحت و هم عدم دقت در مقدار آن نقش دارد. بازهی این شاخص از صفر تا 400 است.
- امتیاز B؛ میانگین روندهی BIS (MRBIS[23]): این شاخص حاصل میانگین گرفتن از ده BIS آخر است و نمایانگر عدمصحت بلندمدت آزمایشگاه است. بازهی این شاخص از 400- تا 400 است.
- امتیاز C؛ انحراف معیار BIS (SDBIS[24]): این شاخص انحراف معیار ده BIS آخر است و نمایانگر میزان پراکندگی اختلافهای نتایج آزمایشگاه است. بازهی این شاخص از صفر تا 442 است. علاوه بر عدمدقت سنجش، عوامل دیگری نیز در این پراکندگی نقش دارند مانند عدمصحت وابسته به غلظت (خطای سامانمند نسبی)، عدمصحت وابسته به زمان (جابجایی کالیبراسیون ناشی از تغییر شماره گروه معرف یا کالیبراتور) و تاثیرات وابسته به نمونه (اثر زمینهای).
یکی از اهداف عمدهی محاسبهی امتیازهای انباشتی از توزیعها و نمونههای متعدد، “ملایم کردن” نوسان طبیعی اختلافها است تا بتوان برآورد مطمئنی از تمایل مرکزی کلی اختلاف و نیز شاخصی از یکدستی اختلاف در طول زمان به دست داد. تمرکز اساسی این برنامه بر اختلاف کلی و یکدستی اختلافها (نوسان) در طول زمان است. باید توجه داشت که وقتی امتیاز مربوط به “یکدستی اختلاف” (نمرهی C) بزرگ باشد، اعتماد به نمرهی اختلاف کلی (نمرهی B) کاهش مییابد (و برعکس).
استفاده از CCV این امکان را نیز فراهم میکند که بتوان با انباشتن نتایج سنجش آنالیتهای گوناگون، شاخصی برای برآورد کلی عملکرد آزمایشگاه محاسبه کرد. این امکان از آن روی فراهم است که CCV هر آنالیت بر اساس یک معیار یکسان یعنی “عملکرد قابل دستیابی با توجه به وضعیت موجود فناوری” تعیین شده است؛ و در نتیجه، اگرچه CCV آنالیتهای گوناگون ارقام متفاوتی است (مثلا 4 برای کلسیم و 18.4 برای CPK) اما همهی آنها نمایانگر یک امر واحد یعنی “کیفیت قابل دستیابی با شرایط و امکانات موجود” هستند و در واقع در برابر یک معیار معین “میزان” شدهاند. با در نظر گرفتن این که در برنامهی UK NEQAS همهی آنالیتها همزمان برای سنجش فرستاده نمیشود و آنالیتهای گوناگون به تدریج و در روزهای مختلف توزیع میشود، از VISهای آخرین 40 سنجش فارغ از نوع آنالیت (نتایج تقریبا تا حدود 6 ماه پیش) میانگین گرفته شده و میانگین روندهی VIS کلی (OMRVIS[25]) حساب میشود. این شاخص میتواند از صفر تا 400 باشد. از این شاخص میتوان به عنوان نشانگر عملی و سودمندی در بررسی کیفیت کلی آزمایشگاه و تغییرات آن استفاده کرد. البته هنگام بررسی باید در نظر داشت که به رغم قابل قبول بودن OMRVIS ممکن است نتایج یک یا چند آنالیت قابل قبول نباشد و بنا بر این نباید بررسی را فقط به ارزیابی OMRVIS محدود کرد.
در این برنامه همچنین نمودارهایی به آزمایشگاه ارائه میشود که در آنها نتایج تا دو سال ونیم پیش آزمایشگاه برای OMRVIS، MRVIS، MRBIS و SDBIS در برابر شمارهی توزیع نمایش داده شده است. در این نمودارها صدکهای 5، 50 و 95 برای OMRVIS و MRVIS مشخص شده است.
برای هورمونها به جای MRBIS و SDBIS به ترتیب شاخصهای BIAS و VAR حساب میشود[26]:
- اختلاف انباشتی؛ BIAS: این شاخص معادل امتیاز B یا MRBIS در بیوشیمی است. برای حساب کردن BIAS نتایج نمونههای قابل استفاده تا 6 ماه پیش، از دادههای پرت پیرایش میشود، سپس اختلاف آن نتایج با مقادیر هدف محاسبه و میانگین ژئومتریک اختلافها حساب میشود.
- نوسان انباشتی اختلاف؛ VAR: این شاخص معادل امتیاز C یا SDBIS در بیوشیمی است. برای حساب کردن VAR نتایج نمونههای قابل استفاده تا 6 ماه پیش، از دادههای پرت پیرایش میشود و سپس انحراف معیار ژئومتریک اختلافها حساب میشود.
منظور از نمونههای قابل استفاده نمونههایی است که به آنها آنالیت خالصشده (مثل استانداردهای بینالمللی) افزوده نشده باشد. نتایج چنین نمونههایی در محاسبات انباشتی وارد نمیشود زیرا احتمالاً این آنالیتها از آنالیت موجود در نمونههای بالینی همگنتر هستند. تفسیر BIAS و VAR مشابه MRBIS و SDBIS است با این تفاوت که چون در مورد هورمونها از CCV استفاده نشده است بنا بر این نتایج آنها در برابر یک معیار یکسان (یعنی کیفیت قابل دستیافت با فناوری موجود) میزان نشدهاند و نمیتوان نتایج آنالیتهای مختلف را ترکیب و شاخص کلی عملکرد مانند OMRVIS حساب کرد.
مرزهای پذیرش برای شاخصهای بیانشده به وسیلهی مجمع مشورتی تضمین کیفیت ملی (NQAAP[27]) پس از تبادل نظر فراوان با سازماندهندگان این برنامه و نیز هیئت مدیره/گروه مشورتی متخصصین (SAG[28]) طوری تعیین میشود که بازتابی از وضعیت کنونی سنجش و نیز مشوقی برای پیشرفت باشد. چنانچه نمرههای شرکتکنندهای در سه توزیع پی در پی بیش از حد مجاز شود عملکرد آن شرکتکننده غیررضایتبخش به شمار خواهد آمد. مجریان UK NEQAS موظف هستند وضعیت شرکتکنندگانی را که به طور پیوسته عملکرد ضعیفی دارند به NQAAP گزارش کند. البته NQAAP به برگزارکنندگان تا حدودی اجازهی انعطافپذیری در تعیین مرزهای پذیرش برای برنامهی هورمون داده است، زیرا در مورد این آنالیتهای ناهمگن، نتایج بیرون از مرزهای پذیرش ممکن است بیشتر بازتابدهندهی اختلافهای وابسته به روش باشد تا عملکرد ضعیف آزمایشگاه.
شیوهی امتیازدهی UK NEQAS این امکان را فراهم میسازد که آزمایشگاه بتواند به آسانی و به شیوهای منطقی و سلسله مراتبی، گزارش عملکرد خود را بررسی کند. آزمایشگاه ابتدا به بررسی OMRVIS میپردازد تا از عملکرد کلی خود در حدود 6 ماه گذشته فارغ از نوع آنالیت آگاه شود. سپس به بررسی MRVISها میپردازد تا آزمایش یا آزمایشهایی را که رد شدهاند شناسایی کند. بررسی MRVIS (به عنوان شاخص خطای کل) این امکان را میدهد که آزمایشگاه آنالیتهایی را که بیشترین مشکل را در رابطه با وضعیت موجود دارند شناسایی کند. در این مرحله باید آزمایشگاه با استفاده از داوری حرفهای و با توجه ویژه به پایش کیفیت داخلی و دیگر اطلاعات، تعیین کند که آیا اقدام اصلاحی لازم است یا نه. در صورت نیاز به اقدام، بررسی MRBIS/BIAS و SDBIS/VAR اطلاعات با ارزشی در بارهی علت ریشهی اشکال فراهم میسازد؛ این شاخصها به ترتیب نشان میدهند که آیا اشکال در افزایش عدمصحت است یا افزایش عدمدقت. شاخص VIS یک شاخص ترکیبی است زیرا هم به عدمصحت و هم به عدمدقت حساس است، اما MRBIS/BIAS و SDBIS/VAR امکان جدا کردن این دو جنبه را فراهم میکند.
در UK NEQAS علاوه بر ارزیابی عملکرد هر آزمایشگاه منفرد، سازماندهندگان برنامه هنگام پردازش نتایج به دقت انسجام و همخوانی نتایج روشهای گوناگون را زیر نظر میگیرند و هرگونه جابجایی غیر منتظرهی وابسته به روش را که ممکن است نشانهی یک جابجایی مهم بالینی باشد موشکافی و با شرکت سازندهی آن روش برای بررسی و اقدامات اصلاحی مطرح میکنند.
نکتهای که باید به آن توجه شود این است که هدف از تعیین CCVها ایجاد معیاری بوده است برای بررسی عملکردی که قابل دسترسی باشد و به هیچ عنوان شاخص کیفیت بالینی مورد نیاز نیست. دلیل حفظ مقادیر آنها از زمان تعیین تا کنون، این بوده است که بتوان تغییرات عملکرد در طول این سالیان را با استفاده از یک معیار ثابت بررسی کرد. برای این که نیازهای بالینی در ارزیابی عملکرد منظور شود، NQAAP برای آنالیتهای گوناگون با توجه به جنبههای بالینی و دیگر جنبههای عملی، ارقام متفاوتی را برای شاخصهای MRBIS/BIAS و SDBIS/VAR به عنوان مرز پذیرش در نظر میگیرد و چنین نیست که بر اساس بازههای آماری برای همهی آنالیتها مرزهای یکسان در نظر گرفته شود؛ مثلا بر اساس ناحیهی فراوانی 95%، اعداد 100 و 200 به ترتیب به عنوان مرزهای عملکرد خیلی خوب و خوب برای همهی آنالیتها باشد. همچنین، همانطور که آمد در سامانهی VI فرض بر ثابت بودن CV در سراسر بازهی سنجش است. اگرچه این فرض در برخی موارد درست نیست، به رغم این، از CV متغیر برای سطوح مختلف غلظت استفاده نمیشود زیرا در آن صورت امکان ارزیابی انباشتی و بلندمدت عملکرد (که هدف اصلی یک برنامهی EQA است) از بین خواهد رفت و در واقع سامانهی امتیازدهی VI به سطح یک سامانهی متغیر مانند سامانهی SDD کاهش خواهد یافت.
همانطور که از مشخصات ارائهشده در بالا برمیآید برنامهی UK NEQAS در مورد آنالیتهایی که با روشهای مرجع ارزشگذاری میشوند یک برنامهی سطح 2 به شمار میآید و در مورد آنالیتهایی که برای آنها از هدف توافقی استفاده میشود یک برنامهی سطح 4 به شمار میآید.
برنامهی EQA در امریکا
فعالیتهای EQA در امریکا در چهارچوب “الزامات بهبود آزمایشگاههای بالینی” (CLIA[29]) انجام میشود. شرکت آزمایشگاهها در برنامههای EQA از سال 1967 و به دنبال تصویب قانون بهبود آزمایشگاههای بالینی (CLIA-67[30]) یک الزام قانونی به شمار میآید؛ اگرچه در سالهای پیش از این قانون، آزمایشگاهها همواره به صورت داوطلبانه در برنامههای EQA که با اهداف آموزشی به وسیلهی تشکلهای حرفهای مانند CAP اجرا میشد شرکت میکردند.
بر اساس طراحی اولیهی CLIA، برگزارکنندگان برنامههای EQA باید در طول سال در سه نوبت برای آزمایشگاهها نمونه بفرستند. در هر توزیع، برای هر آنالیت 5 نمونه در 5 سطح مختلف فرستاده میشود. مرزهای پذیرش در این برنامه در سال 1988 در فهرستی با عنوان CLIA-88 منتشر شده است. در این فهرست، محدودههای پذیرش بیشتر آنالیتها به شکل ثابت یا درصد تعیین شده است، مانند محدودهی ±10% برای کلسترول و ±1 mg/dL برای کلسیم، اما در مورد برخی از آنالیتها، محدودههای پذیرش به صورت آماری و بر اساس انحراف معیار نتایج شرکتکنندگان در هر دور تعیین میشود مانند ±3SD برای TSH. همچنین در این برنامه، میانگین شرکت کنندگان به عنوان مقدار هدف در نظر گرفته شده است.
ارزیابی به این شکل است که در هر توزیع برای هر آنالیت، نتایج سنجشها بسته به این که درون محدودهی مجاز هستند یا نه، به صورت “درست/یا نادرست” معین میشوند. چنانچه در یک توزیع دست کم 4 نتیجه از بین 5 سطح فرستاده شده درست باشد، عملکرد آزمایشگاه برای آن آنالیت در آن توزیع “پذیرفته” به شمار خواهد آمد. برای ارزیابی انباشتی، نتایج سه توزیع آخر بررسی میشود. چنانچه در بین سه توزیع آخر، دو توزیع ناپذیرفتهی متوالی وجود نداشته باشد نتیجهی ارزیابی “رضایتبخش” قلمداد خواهد شد؛ اما چنانچه دو رخداد ناپذیرفتهی پیدرپی وجود داشته باشد عملکرد بلندمدت آزمایشگاه “غیررضایتبخش” عنوان خواهد گرفت. برگزارکنندگان برنامههای EQA موظف هستند عملکرد آزمایشگاهها را به CMS[31] گزارش کنند.
با این اوصاف برنامهی CLIA در طراحی اولیه، به دلیل استفاده از نمونههای تبادلناپذیر و عدم ارسال نمونههای تکراری، یک برنامهی سطح 6 به شمار میآید. همچنین مرزهای پذیرش این برنامه، همانطور که پیشتر گفته شد، بر اساس کیفیت مورد نیاز بالین تعیین نشدهاند بلکه بر اساس الزامات قانونی تعیین شدهاند و نمایانگر حداقل کیفیت لازم برای ادامهی فعالیت یک آزمایشگاه هستند. اما در دو دههی گذشته روند اجرای این برنامه، به ویژه به وسیلهی CAP که یکی از بزرگترین مجریان EQA در امریکاست، تغییرات چشمگیری به سوی یک برنامهی سطح 2 داشته است. در این سالها و به دنبال افزایش آگاهی نسبت به اشکالات نمونههای تبادلناپذیر، مانند مشکلاتی که استفاده از چنین نمونههایی برای آنالیتهای استروئیدی مانند ویتامین D، تستوسترون و استرادیول به دنبال داشت، استفاده از مواد تبادل پذیر رو به افزایش گذاشته است. مقادیر هدف نیز در بسیاری از موارد با بهرهگیری از مواد و/یا روشهای مرجع، روشهای مقایسهای منتخب یا آزمایشگاههای مرجع که از سوی سازمانهایی مانند CDC[32] و [33]NIST در اختیار گذاشته میشوند تعیین میشود. در تعیین محدودههای پذیرش نیز الزامات بالینی بیش از پیش در نظر گرفته میشود مانند مرزهای مجاز در برنامهی CAP.
همچننین در این سالها با افزایش توانمندی برنامههای EQA در امریکا، این برنامهها در فرآیند شناسایی آنالیتهای نیازمند عیارمند/هماهنگ شدن، اجرای برنامههای عیارمندسازی/هماهنگسازی و سپس نظارت بر برقرار ماندن دستاوردهای این اقدامات نقش فراوانی داشتهاند. از برجستهترین این فعالیتها میتوان به عیارمندسازی سنجش کراتینین، A1c، هورمونهای استروئیدی و ویتامین D اشاره کرد (در شکل 4 نمونهی تلاشهای انجام شده برای عیارمندسازی سنجش A1c ارائه شده است). به دنبال موفقیت تلاشهای انجام شده در امریکا برای عیارمند کردن نتایج سنجش ویتامین D، برنامهی DEQAS انگلستان، که یک برنامهی EQA اختصاصی برای ویتامین D است و نمونههایش را به بسیاری از کشورهای جهان میفرستد، از سال 2013 به بعد در برنامهی خود از نمونههای تبادلپذیر که به وسیلهی NIST ارزشگذاری میشوند استفاده میکند.
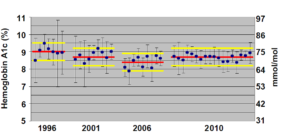
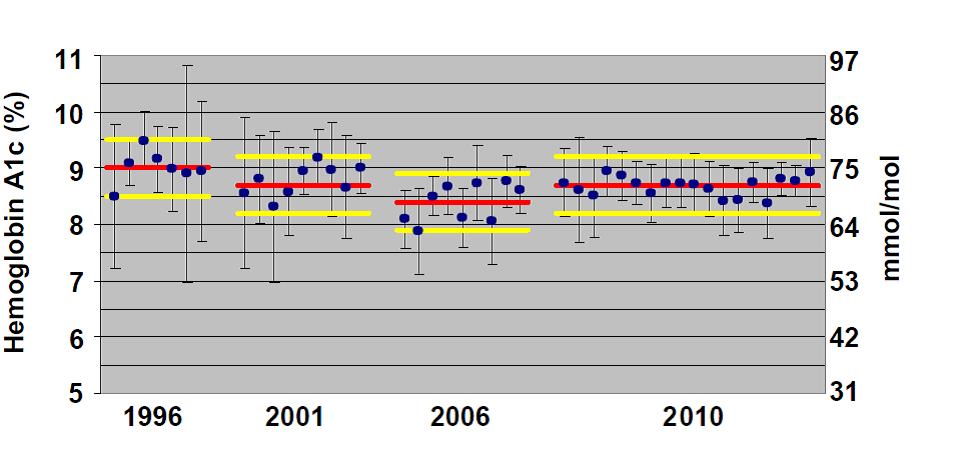
شکل 4 – مثالی از استفاده از نمونهی تبادل پذیر برای نظارت بر پیشرفت در یکدستی نتایج A1c بین روشهای مختلف. ارزیابی CAP-GH2
فقط نتایج نمونهی دارای غلظت میانی از بین نمونههای فرستادهشده در هر توزیع نمایش داده شده است. میانگین نتایج هر گروه به شکل دایرهی آبی توپر و پراکندگی نتایج به صورت میلهی خطا برابر ±2SD دیده میشود. خطهای قرمز نشانگر مقدار هدف هر نمونه است که به وسیلهی آزمایشگاههای مرجع ثانویه در “برنامهی ملی عیارمندسازی گلیکوهموگلوبین” (NGSP[34]) تعیین شدهاند. دو خط زرد رنگ برای هر نمونه نمایانگر مرزهای مجاز ثابت برابر 0.5 واحد فاصله از مقدار هدف است. همانطور که دیده میشود با گذشت زمان، میانگین گروههای بیشتری به مقدار هدف نزدیک شده است. ترجمهی این بهبود عدمصحت همراه با بهبود عدمدقت آن است که میلهی خطای گروههای کمتری از محدودهی مجاز بیرون میزند و این یعنی کاهش خطای کل و دستاورد بهتر برای بیماران. این پیشرفت حاصل تلاش مشترک CAP و بخش مسئول صدور گواهی برای سازندگان در NGSP است.
سخن پایانی
به عنوان جمعبندی و نتیجهگیری، جملات زیر از راهکارنمای WHO در بارهی ارزشمندی برنامههای EQA در سطح کلان کشوری و نیز در سطح خرد برای هر آزمایشگاه ارائه میگردد: “یک برنامهی EQA کارآمد میتواند عیار کلی عملکرد در یک کشور را ارزیابی کند، نیاز به پیشرفت را برانگیزاند و حرکت به سوی همخوانی بهتر بینآزمایشگاهی را زیر نظر بگیرد. چنین برنامهای در همان حال ابزار مدیریتی فوقالعادهای برای آزمایشگاه است، ارزیابی بیطرفانه و مستقلی از عملکرد آزمایشگاه ارائه میکند، پیشرفت را تحریک و پیشروی را پایش میکند. با وجود این، نیل به چنین مقاصدی در گروی طراحی و اجرای درست برنامه است”.
امروزه مسئولیت کیفیت سنجشهای آزمایشگاهی مسئولیتی است مشترک بین آزمایشگاه، آزمایشگاههای مرجع، صنعت مواد تشخیصی و سازمانهای حرفهای. باید همهی این طرفهای سهیم تلاش مشترکی به عمل آورند تا بتوان نتایجی تولید کرد که کیفیت مورد نیاز بالین را تامین کنند. آزمایشگاههای مرجع وظیفه دارند امکان دسترسی به مواد و روشهای مرجع را برای سازندگان و آزمایشگاهها فراهم کنند. صنایع سازندهی مواد تشخیصی باید تلاش کنند روشهایی تولید کنند که در عین برخوداری از پایداری و دقت بالا، از صحت بالایی نیز برخوردار باشند. نیل به چنین مقصودی مستلزم آن است که سازندگان، کالیبراتورهایی را در اختیار آزمایشگاهها بگذارند که رد آنها از طریق یک سلسلهی پیوسته و با عدمقطعیت پایین به مواد و/یا روشهای مرجع میرسد. تنها در این صورت است که سازندگان روشها، فرآیند انتقال کالیبراسیون از مواد و/یا روشهای مرجع به روشهای روزمرهی آزمایشگاهی را به درستی انجام دادهاند. در این راستا، از سال 2003 در اروپا بر اساس قانون IVD Directive 98/79CE سازندگان مواد و وسایل تشخیصی موظف هستند که ردیابیپذیر بودن تولیداتشان را تضمین کنند. سازمانهای حرفهای آزمایشگاهی باید نقش پیشتازانهای در همهی فعالیتهای مربوط به بهبود کیفیت سنجشها از جمله فعالیتهای عیارمندسازی/هماهنگسازی ایفا کنند؛ آن چنان که انجمن شیمی بالینی امریکا (AACC[35]) عمل کرده و از چند سال پیش از پیشتازان هماهنگسازی جهانی نتایج بوده است.
پذیرش جهانی راهکارنماهای اقدامات بالینی مستلزم آن است که ما آزمایشگاهیان نیز جوابهایی دارای یکدستی جهانی تولید کنیم. در صورت اجرای برنامههای EQA توانمند مانند برنامههای سطح 1 و 2، سازمانهای EQA موقعیت ویژهای دارند برای این که از راه شناسایی آزمایشهایی که نیازمند عیارمندسازی/هماهنگسازی هستند و نیز به وسیلهی برانگیختن و برقرار نگهداشتن فعالیتهای عیارمندسازی و هماهنگسازی جهانی، که لازمهی پشتیبانی از راهکارنماهای فعالیتهای بالینی هستند، به شکلی بسیار اساسی بر ارزشمندی عرصهی آزمایشگاهی بیفزایند.
قدردانی:
بر خود لازم میدانم از سرکار خانم نوشین امیری فر دانشجوی PhD دانشگاه روئن فرانسه به خاطر کمک شایان توجه ایشان در تهیهی مقالات و کتب منبع این نوشته کمال قدردانی و تشکر را داشته باشم.
منابع:
- Greg Miller. Proficiency Testing/External Quality Assessment: Current Challenges and Future Directions. Clinical Chemistry 57:12 1670–1680 (2011)
- WHO Guideline. Practice of Quality Assurance in Laboratory Medicine in Developing Countries.
- Aitio, P. Apostolib. Quality assurance in biomarker measurement. Toxicology Letters 77 (1995) 195-204
- Maziotta D, Harel D, Schumann G, Libeer JC. Guidelines for the requirements for the competence of EQAP organizers in medical laboratories. International Federation of Clinical Chemistry (IFCC)/Education and Management Division (EMD)/Committee of Analytical Quality (C-AQ); 2002
- J. R. Healy. Outliers in Clinical Chemistry Quality-Control Schemes. CLIN. CHEM. 25/5, 675-677 (1979)
- Gary L. Horowitz. Proficiency Testing Matters. Clinical Chemistry 59:2 335–337 (2013)
- Andrew Taylor. Quality assessment of measurement. Journal of Trace Elements in Medicine and Biology 25S (2011) S17–S21
- Catharine M. Sturgeon. External quality assessment of hormone determinations. Best Practice & Research Clinical Endocrinology & Metabolism 27 (2013) 803–822
- Perich C, et al, External quality assurance programs as a tool for verifying standardization of measurement procedures: Pilot collaboration in Europe, Clin Chim Acta (2014), http://dx.doi.org/10.1016/j.cca.2013.11.005
- Sturgeon CM, Common decision limits — The need for harmonized immunoassays, Clin Chim Acta (2013), http://dx.doi.org/10.1016/j.cca.2013.11.023
- Jansen R, et al, A category 1 EQA scheme for comparison of laboratory performance and method performance: An international pilot study in the framework of the Calibration 2000 project, Clin Chim Acta (2013), http://dx.doi.org/10.1016/j.cca.2013.11.003
- Braga F, Panteghini M, Verification of in vitro medical diagnostics (IVD) metrological traceability: Responsibilities and strategies…, Clin Chim Acta (2013), http://dx.doi.org/10.1016/j.cca.2013.11.022
- Aarsand AK, Sandberg S, How to achieve harmonization of laboratory testing —The complete picture, Clin Chim Acta (2013), http://dx.doi.org/10.1016/j.cca.2013.12.005
- Henk Baadenhuijsen, et al. Commutability Assessment of Potential Reference Materials Using a Multicenter Split-Patient-Sample Between-Field-Methods (Twin-Study) Design: Study within the Framework of the Dutch Project “Calibration 2000”. Clinical Chemistry 48:9 1520–1525 (2002)
- Christa Cobbaert, et al. Selection, Preparation, and Characterization of Commutable Frozen Human Serum Pools as Potential Secondary Reference Materials for Lipid and Apolipoprotein Measurements: Study within the Framework of the Dutch Project “Calibration 2000”. Clinical Chemistry 48:9 1526–1538 (2002)
- Christa Cobbaert, et al. Systematic monitoring of standardization and harmonization status with commutable EQA-samples—Five year experience from the Netherlands. Clinica Chimica Acta 414 (2012) 234–240
- Birmingham Quality Participants Manual. UK NEQAS website. Updated Thursday, Dec 13, 2012
- Jams O. Westgard. Basic QC Practices. 3rd 2010. Westgard QC, Inc.
- Ravinder J. Sing, et al. (Letter to the editor) Precisely Wrong? Urinary fractionated Metanephrines and Peer-based Laboratory Proficiency Testing. Clinical Chemistry 51:2 1472–1473 (2005)
[1] External Quality Assessment
[2] Proficiency Testing
[3] Standardization
[4] Harmonization
[5] Standard
[6] Commutability
[7] Clinical Laboratory Standards Institute
[8] Traceability
[9] Joint Committee for Traceability in Laboratory Medicine
JCTLM کمیتهی مشترکی است که از سال 2002 با همکاری CIPM IFCC و ILAC برای اجرایی کردن 98/79/EC تشکیل شده است.
[10] Certified Reference Materials
[11] Richtlinien der Bundesärztekammer
[12] Spanish Minimum Consensus
[13] College of American Pathologists
[14] Stichting Kwaliteitsbewaking Medische Laboratorium Diagnostiek
[15] UK National External Assessment Service
[16] Standard Deviation of Differences
[17] Designated Value
[18] Variance Index
[19] Chosen CV
[20] Bias Index Score
[21] Variance Index Score
[22] Mean Running VIS
[23] Mean Running BIS
[24] Standard Deviation of BIS
[25] Overall Mean Running VIS
[26] حروف به کار رفته در ترکیبهای BIAS و VAR مخفف کلمات نیستند و این ترکیبها فقط به عنوان نشانه برای شاخصهای معرفی شده هستند.
[27] National Quality Assurance Advisory Panel
[28] Steering Committee/Specialist Advisory Group
[29] Clinical Laboratory Improvement Amendments
[30] Clinical Laboratory Improvement Act-1967
[31] Center for Medicare and Medicaid Services
[32] Center for Disease Control and Prevention
[33] National Institute of Standards and Technology
[34] National Glycohemoglobin Standardization Program
[35] American Association of Clinical Chemistry
نکتههایی دربارهی QC خالی
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام