
روشهاي شناسايي سریع عوامل میکروبی
دكتر رضا ميرنژاد – باكتريولوژيست، استاديار دانشگاه
شناسايي سريع و دقيق عامل میکروبی جهت درمان افراد مصدوم و كنترل انتقال آلودگي به افراد سالم مهم ميباشد. امروزه در آزمايشگاههاي میکروب شناسی پیشرفته در دنیا جهت تشخيص عوامل میکروبی از روشهاي رايج و سنتي مانند آزمايش مستقيم، كشت و روشهاي ايمنولوژي كمتر استفاده شده و از روشهاي مولكولي مانند (PCR، وسترن بلات، ايمنوبلات، ايمنو اسي، ايمنو فلورسانس و غيره) استفاده ميكنند. روشهای تشخیص مولکولی، جداسازی و شناسائی پاتوژنها را بر اساس شاخصهای مولکولی خاص انجام میدهند. این سیستمها دارای سرعت، حساسیت و اختصاصیت بالا نسبت به روشهای معمول در آزمایشگاه میباشند. همچنین بدلیل اینکه بیشتر سنجشهای مولکولی امروزه، به صورت اتوماتیک قابل دسترس میباشند، لذا نسبت به روشهای سنتی در آزمایشگاه سادهتر انجام میشوند. اصولاً از روشهای مولکولی در آزمایشگاه میکروب شناسی برای شناسائی عوامل غیر قابل کشت (مانند ویروس هپاتیت B)، عوامل با رشد آرام و سختگیر (مانند مایکوباکتریم توبرکلوزیس)، عوامل عفونی که قدرت بیماریزائی بالائی داشته و در کشت دادن امکان آلودگی کارکنان آزمایشگاه وجود دارد (مانند فرانسیسلا تولارنسیس و گونههای بروسلا)، عواملی که در نمونه به مقدار کم وجود دارند (مانند HIV در افراد سرم منفی) و نمونه مورد آزمایش بسیار کم باشد (مایعات مغز- نخاعی و نمونههای جرم شناسی)، متمایز کردن عوامل مشابه از نظر آنتیژنیک، ارگانیسمهای غیرزنده (ارگانیسمهايی که سبب تشکیل کمپلکس ایمن در بدن میشوند) استفاده میشود.
بطورکلی روشهاي شناسايي سریع عوامل میکروبی عبارتند از:
1- جداسازي و شناسايي عامل میکروبی از طريق كشت دادن (معمولاً يك يا دو روز براي بعضي از عوامل طول ميكشد).
2- شناسايي سموم توسط دستگاههاي اسپكتروسكوپي جرمي، تلقيح به حيوانات آزمايشگاهي و يا روشهاي ديگر.
3- تشخيص ايمنولوژيك، شناسايي آنتيباديها (ايمونوگلوبينهاي خاص از نوع IgM كه ممكن است در عرض سه روز پس از عفونت حاصل گردد).
4- شناسايي ضايعات ژنتيكي از طريق شاخصهاي تشخيصی DNA probe
5- شناسايي محصولات متابوليكي عامل عفونتزا و مواد سمي در نمونههاي باليني
تشخیص مولکولی عوامل میکروبی میتواند بر اساس Central dogma بیولوژی مولکولی انجام شود. بر اساس این Central dogma، DNA همانندسازی میکند و RNA از روی DNA نسخه برداری شده و در نهایت RNA نسخه برداری شده (mRNA) در سطح ریبوزوم قرار میگیرد و به پروتئین ترجمه میگردد ( شکل 1). لذا با توجه به این Central dogma اهداف مولکولی در تشخیص میکروبها عبارتند از:
1- DNA: شامل ژنوم کامل- ژن تکی و سکانس کوتاه
2- RNA: به خصوص در RNA ویروسها. این روش خیلی بندرت در باکتریها بکار میرود چرا که RNA در باکتریها پایدار نمیباشد، هر چند در ریبوتایپینگ از RNA استفاده میگردد.
3- پروتئین: شامل پروتئینهای ایمونولوژیک میباشد و اصولاً با روشهای سرولوژی و (Sodium dodecyl sulfate Polyacrylamide gel electrophoresis)SDS-PAGE قابل شناسائی میباشند.
4- دیگر موارد: مانند LPS باکتریها که با روشهای سرولوژی و SDS-PAGE قابل شناسائی میباشد.

شکل 1: dogma مرکزی بیولوژی مولکولی
بطور کلی تکنیکهای مولکولی یا DNA یا RNA را شناسائی كرده و کمتر پروتئینها و مواد دیگر را شناسائی میکنند. امروزه این تکنیکها در آزمایشگاههای تشخیصي و تحقیقاتی بسیار استفاده شده و روزبروز در حال توسعه و تکامل میباشند. تشخیصهای مولکولی بر اساس روش تشخیص به چهار دسته کلی تقسیم میشود:
1- تکنیکهای تشخیصی که نیاز به آمپلی کردن ژن مورد نظر ندارند مستقیماً بدون آمپلی کردن سکانس خاص، میکروب را شناسائی میکنند. مانند In situ hybridization, [1]FISHو غیره.
2-تکنیکهای تشخیصی که ژن مورد نظر را آمپلی میکنند مانند انواع مختلف PCR، [2]TBA و غیره.
3 – تکنیکهای تشخیصی که پروپ نشاندار را آمپلی میکنند مانند :[3]LCR ، PCR/OLA[4] و غیره.
4- تکنیکهای آنالیز و جداسازی بعد از آمپلی کردن مانند ژل الکترفوز، PFGE، کالریمتری، کیمولومینانس و غیره.
در ادامه بحث به جزئيات بعضی از اين روشهاي سريع مولكولي بكار رفته در آزمايشگاههاي مدرن میکروب شناسی جهت شناسايي عوامل میکروبی اشاره خواهد شد.
1- Fluorescence In Situ Hybridization (FISH)
در این روش با کمک شناساگر از نوع 16S -rRNA یا 23S -rRNA نشاندار و میکروسکوپ فلورسانس برای جداسازی مستقیم باکتری از نمونههای کلینیکی مانند خون یا بافت یا بعد از کشت روی محیطهای غنی استفاده میشود (شکل 2). این روش برای جداسازی میکروبهای سخت رشد مانند یرسینا پستیس و بارتونلاها بسیار مفید میباشد و میتوان چندین گونه از باکتریها را به طور همزمان با استفاده از رنگهای فلورسانت مختلف شناسائی کرد. این روش از فیکس کردن نمونه روی لام، هیبرید کردن نمونه با پروپ و بررسی هیبریدها بوسیله میکروسکوپ فلورسانس حدود 1-2 ساعت طول میکشد.
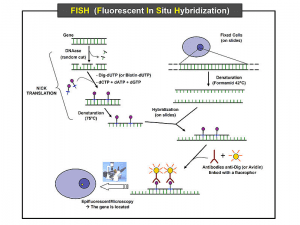
شکل 2: مراحل مختلف تکنیک FISH
2- شناسائی بر اساس سکانس کردن
اگر ارزیابی پاتوژن باکتری خاص مشکل میباشد یا هنگامی که چندین عامل به عنوان عامل بیماری مطرح باشند بایستی از روشی که قدرت شناسائی عوامل در محدوده وسیع را دارند استفاده شود. اهداف عمومی مانند ژنهای16S-rRNA یا نواحی پراکنده ژن 16S –23 S rRNA برای شناسائی باکتریها بسیار مفید میباشد به خصوص که این باکتریها با استفاده از روشهای عمومی مشکل جدا میشوند. سکانس کردن آن 16S -rRNA اغلب برای جداسازی پاتوژنها از نمونههای کشت منفی مورد استفاده قرار میگیرد. امروزه سکانسهای ژن 16S -rRNA برای تایپينگ کردن بعضی از پاتوژنها مورد استفاده قرار میگیرد. این روش بسیار ارزان و سریعتر از روشهای بیوشیمیائی میباشد. میتوان ژنهای 16S -rRNA را با کمک PCR آمپلی کرد و بعد آن را سکانس کرد. لازم بذکر است از اهداف دیگری كه میتوان برای سکانس کردن آن و شناسائی باکتریها مورد استفاده قرار داد پروتئينهای شوک گرمائی (HSP-65 )یا پروتئينهای شوک سرمائی میباشد.
3- الكتروفورز ژل آگارز
الكتروفورز در ژل آگارز در حرارت اتاق در حالت افقي، روشي استاندارد براي جداسازي قطعات DNA است. ژل آگارز در مقايسه با ژل پلي آكريل آميد قدرت تفكيك كمتري داشته ولي محدوده جداسازي بيشتري دارد. قطعات DNA به اندازه 5200جفت باز تا 60 كيلو باز را ميتوان در غلظتهاي متفاوت ژل آگارز از هم جدا نمود. در جدول( 1) مقدار آگارز موردنياز در ژل براي جداسازي ملكولهاي خطي DNA نشان داده شده است. براي ساختن ژل آگارز ابتدا پودر آگارز را در بافر مناسبي كه داراي [5]EDTA است ريخته و حرارت ميدهند تا محلول شفاف و روشني حاصل گردد. اين عمل را ميتوان با گذاشتن مخلوط بافر و آگارز در ماكروويو انجام داد. سپس، محلول را تا 65 درجه سلسیوس سرد كرده و بعد، به درون قالب ژل الكتروفورز يا روي لام ميريزند تا سفت گردد. قبل از ريختن آگارز يك شانه پلاستيكي را روي لام يا درون قالب ژل آگارز قرار ميدهند. اين عمل بدان جهت انجام ميشود تا دندانههاي شانه، چاهکهاي (حفرههاي) كوچكي را در آگارز قبل از سفت شدن تشكيل دهند. براي تعيين فاصله دندانههاي شانه تا كف ظرف قالب ميتوان لامي را در زير دندانههاي شانه قرار داده و پس از تنظيم، لام را برداشته و آگارز را اضافه كرد. زماني كه آگارز سفت شد، ماتريكس ضخيمي را تشكيل ميدهد كه به مقدار غلظت آگارز بستگي دارد. سپس DNA مورد نظر به درون چاهكها وارد ميشود. هنگامي كه جريان برق از ميان ژل عبور كند DNA كه بار منفي در pH خنثي دارد به طرف كاتد حركت ميكند. ميزان مهاجرت DNA در ژل به عوامل زير بستگي دارد:
– اندازه DNA: مولكولهاي بزرگتر، آهستهتر از ملكولهاي كوچكتر در زمان يكسان حركت ميكنند.
– غلظت آگارز: اندازههاي مساوي از ملكولهاي خطي DNA با سرعتهاي متفاوتي در غلظتهاي مختلفي از ژل آگارز حركت ميكنند (جدول1).
– شكل فضائي DNA: ملكولهاي سوپرکویل (ابرمارپيچي) حلقوي و خطي DNA با اندازههاي يكسان در ژل با غلظت مشخص با سرعتهاي متفاوتي حركت ميكنند.
اشكال مختلف به ترتيب متفاوتي حركت ميكنند كه به شرايط موجود بستگي دارد. هنگامي كه ژل سفت شد و نمونهها به آن اضافه گرديد، دستگاه تنظيم برق را وصل كرده تا جريان برق برقرار گردد. ضروري است كه جهت صحيح الكترودها رعايت شود وگرنه، DNA در جهت اشتباه حركت خواهد كرد. توجه شود كه نمونهها در آن قسمتي كه به قطب منفي و بخش مخالف آن به قطب مثبت وصل شده است قرار گيرد.
جدول (1): مقادیر آگارز موردنياز در ژل براي جداسازي ملكولهاي خطي DNA
| مقدار آگارز در ژل (W/V%) | جداسازي ملكولهاي خطي (kbp)DNA |
| 0/3 | 5 تا 60 |
| 0/6 | 1 تا 20 |
| 0/7 | 0/8 تا 10 |
| 0/9 | 0/5 تا 7 |
| 1/2 | 0/4 تا 6 |
| 1/5 | 0/2 تا 3 |
| 2 | 0/1 تا 2 |
نمونه با مقداري از رنگ نشانگر[6] مخلوط ميشود. اين رنگ از دو جزء تشكيل شده كه در دادن اطلاعات تخميني مبني بر حركت نمونه حاوي DNA در ژل مفيد ميباشد. يك جزء يعني سيانول گزيلن (به رنگ سبز) در اندازهاي حدود 4kb حركت ميكند، در صورتي كه جزء ديگر، بروموفنل بلو (رنگ آبي تيره) در اندازهاي حدود 400bp حركت ميكند. از اين رنگها ميتوان به عنوان راهنما براي زمان مناسب انجام آزمايش بر روي ژل استفاده نمود.
پس از انجام عمل الكتروفورز براي مشاهده DNA بايد رنگآميزي ژل با اتیديوم برومايد (EtBR)یا ترکیبات دیگر انجام گيرد. اين رنگ بين بازهاي دو زنجيره DNA نفوذ ميكند و در زير نور اولتراويوله به رنگ قرمز- نارنجي فلورسنس مشاهده ميگردد. مقدار اتیديوم برومايد مورد استفاده به طول قطعه DNA بستگي دارد. بعد از انجام ژل الكتروفورز در محلول اتیديوم برومايد رنگآميزي شده و سپس با آب مقطر شستشو داده ميشود تا رنگ متصل نشده به DNA كه به درون ژل وارد شده حذف گردد. پس از آن از ژل توسط دستگاهGel Doucomentation
عكسبرداري ميشود (شکل 3).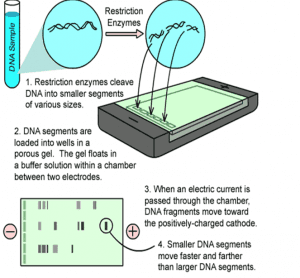
شکل3: مراحل مختلف الکتروفورز
4- Pulsed Field Gel Electrophoresis (PFGE)
تمام ملكولهاي زنجير مضاعف DNA خطي تا اندازه معيني به يك ميزان از ميان ژل آگارز عبور ميكنند. بالاتر از اين حد معين سرعت عبور ملكولهاي DNA از ميان ژل آگارز به اندازه DNA بستگي نداشته و به طور عمده به شدت جريان الكتريكي بستگي دارد. در عمل اين ارتباط بدان معني است كه DNAهای بزرگتر از 40kb نميتوانند با استفاده از جريان ثابت الكتريكي در ژل آگارز افقي به سهولت از هم جدا شوند. اين مشكل را ميتوان با PFGE حل نمود كه در آن، جريان الكتريكي به طور متناوب در دو جهت مختلف با زمانهاي pulse از 1/0 تا 1000 ثانيه يا بيشتر تغيير ميكند (شکل 4). با اين روش ميتوان ملكولهاي DNA موجود در نمونههاي مشكوك تا حدود 5 ميليون باز درازا را از هم جدا كرد. در اين حالت زماني كه ملكول DNA نياز دارد تا مسير خود را در پاسخ به نوسان جريان الكتريكي تغيير دهد به اندازه ملكول DNA بستگي دارد، يعني ملكولهاي كوچكتر با سرعت بيشتري ميتوانند تغيير جهت داده و بنابراين سريعتر از ملكولهاي درازتر از ميان ژل عبور میكنند (شکل 5).

شکل 4: جهات حرکت قطعات اسید نوکلئیک در PFGE
نكات زير در انجام PFGE اهميت بسيار داشته و بايد به آنها توجه شود:
1- فضاي جداسازي: در اكثر سيستمهاي PFGE، DNA در فضاي كوچكي الكتروفورز ميشود و وضوح آن به پيچيدگي نمونه بستگي دارد.
2- قدرت ميدان الكتريكي: قدرت ميدان الكتريكي تأثير مهمي بر جداسازي در روش PFGF دارد، به طوري كه بين زمان جداسازي و مشاهده واضح DNA به اندازههاي خاص به ميدان الكتريكي خاصي نياز ميباشد.
3- زمان Pulse: اولين تأثير زمان pulse، تغيير ميزان جداسازي است. از زمانهاي pulse طولانيتر براي جداسازي DNAهاي بزرگتر استفاده ميشود.
4- زاويههاي تغيير جهت: جداسازي DNA در زاويههاي 165-95 درجه برابر و مشابه است. با اين حال، هر قدر زاويه كوچكتر باشد، حركت DNA سريعتر ميباشد. براي جداسازي DNAهاي خيلي بزرگتر، زاويههاي 105-96 درجه مورد نياز است تا جداسازي DNA در كوتاهترين زمان ممكن به خوبي انجام شود.
5- بافر: از بافرهاي TBE و TBE استفاده ميشود.
6- آگارز: غلظت و نوع آگارز در جداسازي DNA مؤثر است. سريعترين حركت و بهترين وضوح در ژلهايي بدست ميآيد كه از Low electroendosomsis ساخته شده باشد. البته آگارز الكتروفورز معمولي براي PFGE مناسب است.
7- دما: مناسبترين درجه حرارت براي انجام PFGE دماي 14 درجه سلسیوس است.
كاربردهاي PFGE و الكتروفورز ژل آگارز
اصولاً الكتروفورز معمولي و PFGE براي جداسازي و تعيين اندازه پلاسميدهاي بزرگ و DNAهاي كروموزومي باكتريهاي موجود در نمونههاي مشكوك به عوامل میکروبی استفاده ميشود. پس از الكتروفورز كردن نمونههاي مشكوك و بدست آوردن باندهاي مناسب، آنها با الگوهاي مشخص مقايسه ميشوند و عامل بيولوژيك تشخيص داده ميشود.
از كاربردهاي رايج ديگر الكتروفورز ژل آگارز و به خصوص PFGE در ميكروب شناسي پزشكي، مقايسه اپيدميولوژي باكتريهايي است كه متعلق به يك خانواده هستند و در عفونتهاي بيمارستاني و يا به دليل تماسهاي مستقيم انتقال مييابند. مقايسه باندهاي مشاهده شده ممكن است دخالت سويههاي جدا شده در اپيدميها را نشان دهد.

شکل 5: مراحل مختلف PFGE
5- الكتروفورز ژل پلي اكريل آميد- سديم دودسيل سولفات (SDS-PAGE)
براي آناليز يك پروتئين خاص براي شناسايي يك عامل بيولوژيك (به خصوص سموم) در نمونههاي مشكوك از الكتروفورز ژل پلي اكريل آميد استفاده ميشود. در اين روش، نمونه حاوي پروتئين مورد نظر در شرايطي قرار ميگيرد كه پروتئينها به زير واحدهاي پليپپتيدي از هم تفكيك ميشوند و غلظت پليپپتيدهاي متفاوت در مخلوط كاهش مييابد. بعد از مراحل تهيه، مخلوط پروتئيني بوسيله ژل پلي اكريل آميد، الكتروفورز ميشود. سپس ژل را براي مشاهده پروتئينها، رنگآميزي كرده و براي آناليزهاي بعدي مورد استفاده قرار ميگيرد.
پروتئين مورد نظر بخشي از مخلوطي از پروتئينهاي متفاوت است. براي اطمينان از جدا شدن پروتئينها به زير واحدهاي پليپپتيدي و كاهش تجمع، معمولاً پروتئينهاي موجود در مخلوط را تجزيه كرده، كاهش داده و يك دترجنت آنيوني (يعني دودسيل سولفات سديم ] SDS قليائي[) به آن اضافه ميشود. عمل تجزيه شدن نمونه را ميتوان با جوشاندن به مدت كوتاه انجام داد. اين عمل، ساختمانهاي دومي، سومي و چهارتايي را كه ممكن است وجود داشته باشد و همچنين معكوس شدن واكنشهاي تصادفي پروتئين– پروتئين را كه ممكن است رخ دهد تخريب ميكند. عمل كاهش را ميتوان با افزودن بتامركاپتواتانول (BME) انجام داد. اين معرف اتصالهاي دي سولفيدي را با تخريب هر نوع ساختمان سومي كه ممكن است موجود باشد حذف ميكند و هم چنين مانع تشكيل هرگونه اتصالهايي ميشود كه ممكن است پليپپتيدهاي cross-link را از هم مجزا سازد.
افزودن SDS به پليپيتيد تخريب شده بار خالص منفي را به آن اعطا ميكند. مقدار اتصال SDS به پروتئين، نسبت خطي با اندازه پروتئين دارد و به توالي اسيد آمينه بستگي ندارد. چون SDS، بار خيلي بالايي دارد، پس به طور گستردهاي اتصال مييابد و هر بار ديگر موجود در روي پروتئين ناديده گرفته ميشود. بدين ترتيب، بار هر واحد وزن، پايدار بوده و حركت در الكتروفورز به وزن مولكولي آن وابسته است.
این ژل با جوشاندن مخلوط پروتئين در بافري كه حاوي BME،SDS ميباشد حاصل ميگردد. اين امر منجر به ايجاد بار منفي در مخلوطي از كمپلكسهاي پروتئين– SDS ميشود. بافر همچنين داراي رنگ نشانگر ميباشد كه نشان دهنده مساحت حركت انجام شده در روي ژل الكتروفورز ميباشد. گلیسرول، ماده ديگري از بافر است كه درload کردن نمونه به درون ژل كمك ميكند. پروتئينهاي موجود در مخلوط با عبور نمونه از ميان شبكه آكريل آميد از هم جدا ميگردند. اين شبكه به عنوان غربالي، عبور پروتئينها را با حركت آهسته فيزيكي آنها به تأخير مياندازد. مولكولهاي بزرگتر، آهستهتر از مولكولهاي كوچكتر حركت ميكنند. مقدار درصد آكريل آميد در ژل همچنين بر روي ميزان حركت پروتئينها اثر دارد. محلول غليظ پروتئينها، آهستهتر از غلظتهاي پائين حركت ميكنند. در جدول(2) ميزان خطي جدايي پروتئينها با غلظتهاي متفاوت آكريل آميد نشان داده شده است.
جدول (2 ): نسبت غلظت آكريل آميد و جدا شدن پروتئينها
| غلظت آكريل آميد (%) | حدود جدا شدن خطي (كيلودالتون) |
| 15 | 12-43 |
| 10 | 16-68 |
| 7/5 | 36-94 |
| 5 | 57-212 |
پليپپتیدهاي جدا شده توسطSDS-PAGE را ميتوان با استفاده از رنگهاي شيميايي مشاهده كرد. پليپپتيدها را ميتوان به طور همزمان با مخلوطي از اسيد استيك و متانول و رنگآميزي كوماسي برلیانت بلو[7] تثبيت نمود.
اين رنگ براي برخي از اسيدهاي آمينه (براي مثال، آرژينين و لیزین) و رنگآميزي پليپپتيدها در درون ژل اختصاصي است. رنگ اختصاصي را ميتوان با رنگبري به مدت طولاني از ژل خارج كرد. با مقايسه باندهاي حاصل با باندهاي استاندارد شده در روي ژل (پروتئينهايي با اندازه شناخته شده) ميتوان وزن مولكولي پليپپتيد مورد نظر را تخمين زد. عمل الكتروفورز بوسيله ژل با استفاده از ماركرهايي با وزن مولكولي مشخص كه از قبل رنگآميزي شده، تسهيل شده است، به طوري كه مشاهده پايان عمل، قبل از اينكه پروتئين مورد نظر از ژل خارج شود، مقدور ميباشد.
[1] -Fluorescence In Situ Hybridization(FISH)
[2] – Transcription Basel Amplification (TBA)
[3] – Ligase Chain Reaction(LCR)
[4] -PCR/ oligonucleotide legation assay
[5] -Ethyl enediamine tetraacetic acid
[6] – loading buffer
[7] – Comassie brilliant blue
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام