
گزارش موردی
ترشح شیر مستمر و هیپرکلسمی عودکننده
دکتر سید رضا موسوی
شرححال:
بیمار خانمی است 18 ساله با ترشح شیر دوطرفه مستمر، افزایش وزن و هیپرکلسمی عودکننده. در 13 سالگی و قبل از شروع قاعدگی ترشحات شیر از هر دو پستان او مشاهده میشد. این ترشحات همچنان ادامه داشت تا اینکه در سن 16 سالگی سردردهای مکرر و تغییرات دامنه دید نیز به آن اضافه شد. مقدار پرولاکتین بالاتر از ng/ml 2400 بود. سال بعد ماکروآدنوم هیپوفیز او با جراحی برداشته شد. بعد از جراحی دچار کمکاری همهجانبه هیپوفیز و نیز دیابت بیمزه مرکزی(DI) شد. برای جبران کمکاری تیروئید ثانویه به وی لووتیروکسین (T4) داده شد و برای جبران سایر نارساییهای هورمونی،
desmopressin accetate (آنالوگ صناعی ADH)، استروژن کونژوگه، هورمون رشد و برموکریپتین تجویز گردید. سال بعد دچار سنگهای کلیه مکرر شد و تشخیص هیپرپاراتیروئیدیسم اولیه محرز گردید. همچنین ندولهای متعدد تیروئید در وی مشاهده شد. طی یک عمل جراحی هر چهار غده پاراتیروئید و کل غده تیروئید برداشته شد. پس از آن یک هیپوپاراتیروئیدیسم موقت مشاهده شد و متعاقب آن عود هیپرپاراتیروئیدی اولیه به همراه نفروکلسینوز و پیلونفریت ظاهر گردید.
| سؤالاتی که باید مدنظر باشد:
1- مجموعه نتایج آزمایشگاهی و علائم بالینی چه تشخیصی را پیشنهاد میکند؟ 2- چه اختلالات دیگری ممکن است در این بیماری اولیه ظهور پیدا کند؟ 3- چه آزمایش ژنتیکی در این جا کاربرد دارد؟ |
وی برای بررسی بیشتر در بیمارستان بستری شد. وزن وی زیاد شده و دچار افسردگی و اضطراب بود. همچنین از درد مفاصل، خستگی، تاری دید، کاهش اشتها، دیسپنه، پلیاوری و بیخوابی شکایت داشت.
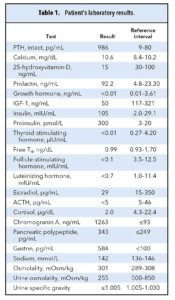
اسهال گاهوبیگاه، بدون نفخ قابلتوجه نیز از دیگر موارد شکایت بود. هیچ سابقه خانوادگی بیماریهای اندوکرین وجود نداشت. در معاینه فیزیکی چاقی، بدحالی، تورم پا و گام برداشتن نامتوازن مشهود بود. ضربان قلب 91 و فشارخون 113/67 میلیمتر جیوه بود. در معاینات قلبی عروقی و تنفسی نکته قابلتوجهی وجود نداشت. پلاکها و ندولهای متعددی در تنه و شکم و اطراف واژن و کشاله ران دیده میشد. بررسیهای آزمایشگاهی (جدول 1)، تصویربرداری و هیستولوژیک بعمل آمد.
در تست تحریک ACTH ( cosyntropin µg 1) غلظت پیک کورتیزول را µg/ dL18/7 نشان داد (محدوده مرجع بزرگتر از 20µg/ dL). در آزمایش CBC، آنمی میکروسیتیک هیپوکرومیک دیده میشد.
بحث
در تصویربرداری MIR تومور هیپوفیز عودکننده و پیشرونده مشاهده گردید. بررسی هیستولوژیک برش تومور هیپوفیز وجود آدنوما را که با ایمونوهیستوشیمی پرولاکتین مثبت بود، تأئید کرد. سونوگرافی اندوسکوپیک کیستهای متعددی را به ابعادmm 4 یا کمتر در پانکراس نشان داد. یک ناحیه ندولار به ابعاد 6/3×4/6 میلیمتر در بدنه پانکراس مشاهده شد که تومور نورواندوکرین پانکراس را پیشنهاد میکرد. بیوپسی با سوزن نازک از این ضایعه بعمل آمد که در آزمایش بافتشناسی تومور نوروآندوکرین پانکراس با سلولهای آتیپیک با گرید کم و متوسط را تأیید کرد. شرححال بیمار، یافتههای معاینات فیزیکی، نتایج آزمایشگاهی، تصویربرداری و پاتولوژی در مجـموع type 1 (MEN1) multiple endocrine neoplasia به همراه آدنوم هیپوفیز، هیپرپاراتیروئیدیسم اولیه و تومورهای نورواندوکرین پانکراس را پیشنهاد میکرد. از نظر وجود موتاسیون ژن (MEN1)آزمایش بعمل آمد که نتیجه مثبت بود. وضعیت بالینی او بهواسطه نارسایی مرکزی آدرنال، هیپوتیروئیدیسم، هیپوگونادیسم و نارسایی هورمون رشد پیچیده شده بود. این موارد به دنبال جراحی ماکروآدنوم هیپوفیز رخ نموده بودند.
پایین بودن کورتیزول و نیز پایین بودن نامتناسب ACTH علیرغم هیپوکورتیزولیسم، نارسایی مرکزی آدرنال را پیشنهاد مینمود. تست تحریک ACTH که به آن تست cosyntropin هم میگویند به افتراق نارسایی اولیه و ثانویه آدرنال کمک میکند. در نارسایی ثانویه آدرنال پاسخ به cosyntropin معمولاً کند است ولی گاهی در حالی که مقدار پایهACTH در محدوده مرجع یا پایینتر است، پاسخ نرمال مشاهده میشود.
آزمایش با دوز µg 250 کوزینتروپین به منظور آزمایش حداکثر ظرفیت آدرنوکرورتیکال است. در این دوز بالا اگر ترشح کورتیزول نسبتاً کم باشد مشخص نمیگردد. آزمایش با دوز µg1 کوزینتروپین حساستر و دقیقتر از دوز µg250 در تشخیص نارسایی نسبی آدرنال میباشد، بهویژه در مواردی که نارسایی ثانویه بهواسطه تومور هیپوفیز یا درمان طولانی با گلوکوکورتیکوئیدها باشد. تست تحریک مستقیم هورمون آزادکننده کورتیکوتروپینها به تشخیص نارسایی ثانویه آدرنال کمک میکند. اگرچه تومورهای تیروئید در مبتلایان به MEN1 دیده میشود ولی پیشنهاد شده است که ناهنجاری تیروئید ممکن است تصادفی بوده و مهم نباشد. در این بیمار تومورهای نورواندوکرین پانکراس گاسترینوما (سندرم زولینگر- الیسون) و انسولینوما معرف تیپیک MEN1 هستند. این بیمار همچنان تظاهرات پوستی MEN1 نظیر آنژیوفیبروما را دارد.
MEN1 یک سندرم ارثی اتوزومال غالب است که با وقوع تومور در 2 یا بیشتر از غدد اندوکرین اصلی مانند تیروئید، بافت اندوکرین پانکراس و هیپوفیز مشخص میشود (2،1). سایر نئوپلاسمهای اندوکرین و غیراندوکرین در MEN1 شامل تومورهای کارسینوئید، تومورهای اندوکرین برونشیال، تومورهای جلدی (آنژیوفیبروم، کولاژنوما، لیپوما، ملانوما)، تومورهای سیستم اعصاب مرکزی (مننژیوما، اپندیمونا، شوآنوما) و تومورهای عضلات صاف (3) میباشند. بیشتر تومورها در MEN1 خوشخیم هستند اما بندرت تومور بدخیم هم دیده میشود. شیوع این بیماری مشخص نیست اما بر اساس بیوپسیهای بعمل آمده وقوع آن 0/25% تخمین زده میشود (2). علت ایجاد MEN1 جهش در ژن MEN1 است که بر روی کروموزوم (11q13) قرار داشته و دارای 10 اگزون است و یک پروتئین هستهای را مشتمل بر 610 اسیدآمینه کد میکند. نام این پروتئین menin است (5،4،1). تصور میشود که menin در فرآیندهای سلولی مهم نظیر تنظیم سیکل سلولی، کنترل نسخهبرداری، تقسیم سلولی و پایداری ژنومی نقش دارد ولی نقش دقیق آن هنوز مشخص نشده است. تناسبی بین ژنوتیپ و فنوتیپ MEN1 مشاهده نشده است (1). در مورد این بیمار چون هیچگونه سابقه خانوادگی ابتلا به اختلالات اندوکرین وجود ندارد این تظاهرات بخاطر موتاسیون اولیه باید باشد. وقوع موتاسیون اولیه در 10% کل موارد MEN1 صورت میپذیرد (2).
تشخیص MEN1 به یکی از صور ذیل محقق میگردد:
- وجود 2 یا بیشتر تومور اندوکرین
- وجود تومور وابسته به MEN1 در یکی از بستگان درجه اول شخص که علائم بالینی MEN1 را دارد.
- تشخیص موتاسیون MEN1 در germline فرد بدون علامت که هنوز علائم آزمایشگاهی یا رادیولوژیک دال بر وجود تومور را بروز نداده است (6،2). تشخیص MEN1 تا چندین سال طول میکشد (6). عدمتشخیص تظاهرات بالینی چندگانه MEN1 علت معمول در تأخیر تشخیص است. هیپرپاراتیروئیدیسم اولیه متداولترین و معمولاً اولین اختلال بالینی در MEN1 است (95%)، پس از آن تومورهای اندوکرین پانکراس (70%-40%) و آدنوما (40%-30%) معمول هستند (7،2). در تمام افرادی که مشکوک به MEN1 هستند غلظت کلسیم و PTH سرم باید اندازهگیری شود. سایر آزمایشهای بیوشیمی مانند انسولین، پروانسولین، گلوکاگون، پرولاکتین، گاسترین، IGF-1 پلیپپتید پانکراس و Chromagranin A در رسیدن به تشخیص میتوانند کمک کنند (8). در افراد مشکوک به MEN1 حتی در صورت عدم وجود علائم بالینی، آزمایش موتاسیون ژنتیکی توصیه میگردد (9،2،1). تصویربرداری با اشعه ایکس و اسکن هیپوفیز، تیروئید و شکم در ابتدای تشخیص و پس از آن بهطور دورهای برای پایش تومور یا در صورت تشدید علائم لازم است. شیوع MEN1 بین یک تا ده نفر در هر 100000 نفر تخمین زده میشود و در اکثر موارد تومورها خوشخیم هستند اما خود بیماری خوشخیم نیست و میتواند کشنده باشد، بنابراین تشخیص بههنگام در کسانی که هنوز علائم بالینی نشان ندادهاند خیلی مهم است. در هر فرد مشکوک به MEN1، تاریخچه خانوادگی کامل، معاینه و آزمایشهای بیوشیمیایی، پاتولوژیک و تصویربرداری ضرورت دارد. مداوای تومور در مبتلایان MEN1 شبیه مداوای آن تومور در افراد غیر MEN1 است، اما بهواسطه پیچیدگی و درگیری اعضای مختلف، مداوای تومورهای MEN1 بخوبی تومورهای دیگر امکانپذیر نیست (2).
گرچه بعد از جراحی وسیع پاراتیروئید، هیپوپاراتیروئیدیسم رخ میدهد، اما وقوع هیپرپاراتیروئیدیسم عودکننده هم در این موارد بعید نیست. بیمارانی که پاراتیروئیدکتومی سابتوتال موفق داشتهاند اغلب در طی 15 سال با عود هیپرپاراتیروئیدیسم مواجه میگردند (10،1). در این بیمار دیابت بیمزه مرکزی به علت تخریب هیپوتالاموس یا هیپوفیز در طی عمل جراحی ایجاد شده است. ویژگی این بیماری افزایش حجم ادرار و کاهش اسمولالیته و وزن مخصوص ادرار بهواسطه کاهش ترشح ADH است. کلسیم پلاسما اندکی افزایش دارد. کمبود ویتامین D شدت هیپرکلسمی را کاهش میدهد اما افزایش PTH را تشدید میکند. بیمار همچنین برای جبران کمبود ویتامین D ارگوکلسیفرول مصرف میکند. رفلاکس اسید معده که در نتیجه گاسترینوما ایجاد شده است بخوبی به امپرازول جواب میدهد. باید توجه داشت که امپرازول بطور کاذب موجب افزایش Chromogranin A میگردد. این ماده یک شاخص مفید در تشخیص نئوپلاسمهای اندوکرین نظیر کارسینوئیدها، نوروبلاستوما، کارسینومای مدولار تیروئید، بعضی تومورهای هیپوفیز و تومورهای فعال و غیرفعال آیلت سلها است. اثر امپرازول در افزایش Chromogranin A متناسب با دوز و طول دوره درمان میباشد.
| نکات مهم:
– MEN1 یک اختلال ارثی اتوزومال غالب است. وجه مشخصه آن وجود 2 یا بیشتر از تومورهای اندوکرین در غدد پاراتیروئید، سلولهای آیلت پانکراس و هیپوفیز است که ممکن است به همراه سایر نئوپلاسمهای اندوکرین باشد. هیپرپاراتیروئیدیسم اولیه متداولترین علامت MEN1 است. – موتاسیونهای MEN1 و سایر ژنهای همراه با MEN1 شناسایی شدهاند. آزمایش موتاسیون MEN1 germline برای غربالگری و تشخیص MEN1 توصیه میشود. – در افراد مشکوک به MEN1، کلسیم سرم و PTH باید اندازهگیری شود. سایر آزمایشهایی که فعالیت اعضای اندوکرین را بررسی میکنند به همراه تصویربرداری در تشخیص بموقع MEN1 مفید هستند. – تشخیص MEN1 بهوسیله خصوصیات بالینی، ارثی یا ژنتیک و یا مجموعهای از این خصوصیات ممکن میگردد. |
این مقاله ترجمهای است از :
A Patient with Persistent Lactation and Recurrent Hypercalcemia
Qing H. Meng and Elizabeth A. Wagar
Clinical Chemistry 61:11
1328–1332 (2015)
اختلالات غده آدرنال (1) سندرم کوشینگ
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام