تشخیص آزمایشگاهی و درمان اختلالات مادرزادی فیبرینوژن
(بخش اول)
اکبر درگلاله1، حسن مروتی2
1- گروه هماتولوژی و طب انتقال خون، دانشگاه علوم پزشکی ایران
2- عضو هیئت علمی مرکز تحقیقات واکسن و سرمسازی رازی
مقدمه
اختلالات مادرزادی فیبرینوژنCFD) [1])، اختلالات انعقادی هستند که با الگوی اتوزوم غالب یا اتوزوم مغلوب به ارث میرسند. این اختلالات شامل چهار زیرگروه آفیبرینوژنمی (با سطوح غیرقابل تشخیص آنتیژن و فعالیت فیبرینوژن)، هیپوفیبرینوژنمی (با کاهش سطوح آنتیژن و فعالیت فیبرینوژن)، دیس فیبرینوژنمی (با سطوح نرمال آنتیژن همراه با کاهش فعالیت فیبرینوژن) و هیپودیس فیبرینوژنمی (با سطوح کاهشیافته آنتیژن همراه با کاهش نامتناسب عملکرد فیبرینوژن) هستند (1). با وجود اینکه شیوع این اختلالات بهصورت کاملاً دقیق مشخص نیست، اما طبق گزارش بانکهای اطلاعاتی بینالمللی، ۸٪ اختلالات انعقادی نادر را شامل میشوند. با این حال، به نظر میرسد که تعداد بیماران فاقد علائم بالینی، کمتر از تعداد حقیقی آنها گزارش شده است. آفیبرینوژنمی با شیوع یک در هر یک میلیون نفر در کشورهایی با ازدواجهای فامیلی بالا، شیوع بالاتری دارد (4-2)؛ اما به نظر میرسد شیوع سایر زیرگروهها، به دلیل تعداد بیشتر بیماران بدون علائم بالینی، بالاتر باشد (5). علائم بالینی این اختلالات به زیرگروه بیماری و سطح فیبرینوژن بستگی دارد (6). تشخیص اولیه بر مبنای تستهای آزمایشگاهی روتین صورت گرفته و با تعیین ژنوتایپ تأیید میشود (3). در مراکز تخصصیتر، بررسیهای عملکردی بیشتر برای تعیین هر چه بهتر فنوتیپ بیماران انجام میشود (7). مدیریت و درمان بیماران اغلب چالش برانگیز بوده و به دلیل فقدان کارآزماییهای بالینی تصادفی یا مطالعات مشاهدهای بزرگ، عمدتاً بر اساس توصیههای موردی و متخصصین است (3,8). در این فصل، بعد از یک نگاه کلی به ساختار فیبرینوژن، به بررسی علائم بالینی، استراتژی تشخیص و مدیریت این اختلالات خواهیم پرداخت.
ساختار و عملکرد فیبرینوژن
فیبرینوژن، پروتئینی با وزن ملکولی بالا (۳۴۰ کیلودالتون) و نیمهعمر حدوداً ۴-۲ روز است که عمدتاً در کبد تولید میشود. علاوه بر این، این پروتئین در سلولهای اپیتلیال روده (9)، اپیتلیال سرویکس (10) و اپیتلیال ریه (11) نیز تولید میشود. مقدار کمی از فیبرینوژن در گردش بهوسیله گرانولهای آلفای مگاکاریوستها و پلاکتها جذب و مجدداً بعد از آزادسازی گرانولهای پلاکت، وارد جریان خون میشود (13, 12). فیبرینوژن یک پروتئین فاز حاد بوده که میزان آن در بارداری، التهاب، عفونت و بدخیمیها افزایش مییابد. این پروتئین، یک ملکول هگزامر با پیوندهای دیسولفیدی است که از دو زیر واحد همولوگ تشکیل شده است. هر زیر واحد شامل یک زنجیره Aα، یک زنجیره Bβ و یک زنجیره γ با وزن ملکولی به ترتیب ۶۴، ۵۷ و ۴۷ کیلودالتون و تعداد ۶۱۰، ۴۶۱ و ۴۱۱ اسیدآمینه است (16-14). زنجیرههای فیبرینوژن به ترتیب بهوسیله سه ژن مجزای FGB، FGA و FGG کد میشوند. این ژنها در حدفاصل سانترومر تا تلومر قرار گرفتهاند و ناحیهای به طول kb 50 در جایگاه ژنی 4q32 را پوشش میدهند. FGA و FGG از روی رشته معکوس و در جهت مخالف FGB رونویسی میشوند. بیان ژن فیبرینوژن بهوسیله فعالیت پروموترها و توالیهای تقویتکننده کنترل میشود (17). ژنهای FGB، FGA و FGG به ترتیب از ۶ (kb 7/5)، ۸ (kb 8) و ۱۰ اگزون (kb 8/5) تشکیل شدهاند (20-18). طی آلترناتیو اسپلایسینگ[2]، دو ایزوفرم برای زنجیره α و دو ایزوفرم برای زنجیره γ ایجاد میشود. رونوشت زنجیره Aα که رونوشت اصلی ژن است توسط اگزون ۵-۱ ژن FGA کد میشود، در حالی که ایزوفرم E-Aα، توسط اگزون ۶-۱ کد میشود که حدود ۲-۱ درصد رونوشتها را به خود اختصاص میدهد. از طرفی رونوشت اصلی ژن FGG، دارای ۱۰ اگزون بوده و مسئول کد کردن زنجیره متداول Aγ است و ایزوفرم فرعی آن (′γ) شایع نبوده و ۲۰ آمینواسید منحصر به فرد را در انتهای کربوکسیلیک زنجیره g کد میکند (21). ژن FGB تنها یک رونوشت mRNA متمایز به طول kb1/9 کد میکند (24-22). تغییرات پس از ترجمه[3] بهصورت N گلیکوزیلاسیون، هیدروکسیلاسیون، اکسیداسیون، دآمیناسیون، سولفاسیون، سیالیشن[4] و فسفوریلاسیون بر روی ملکول فیبرینوژن صورت میگیرد، همچنین بعد از ترجمه، سرهمبندی فیبرینوژن در شبکه آندوپلاسمی خشن، با برقراری پیوندهای دیسولفیدی بهوسیله ملکولهای چاپرون صورت می گیرد (26، 25).
فیبرینوژن دارای ساختار ندولار سهتائی[5] شامل یک ناحیه مرکزی E و دو ناحیه انتهایی D است که هر کدام از چندین دامین ساختاری تشکیل شدهاند (شکل ۱-۶) (27). انتهای آمینی هر شش زنجیره، بخش مرکزی ناحیه E را تشکیل می دهد (28، 16، 15). ناحیه D توسط انتهای کربوکسیلیک زنجیرههای Bβ و γ شکل میگیرد. پایانه کربوکسیل زنجیره Aα (Cα) انعطافپذیر بوده و تمایل به گسترش در نزدیکی ناحیه E را دارد. فیبرینوژن محلول در حضور کلسیم و ترومبین به فیبرین نامحلول تبدیل میشود. در این روند، در آغاز، فیبرینوپپتید A (FPA) و B (FPB) به ترتیب در جایگاه AαArg16-Gly17 و BβArg14-Gly15 توسط ترومبین برش داده میشود که منجر به تشکیل منومر فیبرین (Fragment X) میشود (31-29). فیبرینوپپتیدA سریعتر و زودتر از فیبرینوپپتید B آزاد میشود و برای پلیمریزاسیون فیبرین کافی است. بعد از تبدیل فیبرینوژن محلول به فیبرین نامحلول، لخته فیبرینی بهوسیله فعالیت ترانسگلوتامینازی فاکتور XIII در حضور کلسیم پایدار میشود. فاکتور XIII بین نواحی E و D منومرهای فیبرین مجاور هم، پیوند کووالانسی برقرار میکند و پروتوفیبریل تشکیل میشود، بهصورتی که گلوتامیکاسید دامین D یا E به لیزین دامین مجاور متصل میگردد. در این فرایند هر دو زنجیره Aα و زنجیره γ شرکت میکنند (34-32). فیبرینوژن علاوه بر نقش شناختهشده آن در هموستاز یعنی حمایت از تجمع پلاکتی و تشکیل فیبرین، در
بهبود زخم، رگزایی، تشکیل تومور، تکثیر و مهاجرت سلولی، چسبندگی سلولی، تعاملات ماتریکس سلولی با اتصال به فیبرونکتین و حفظ بارداری نیز نقش دارد (41-35).
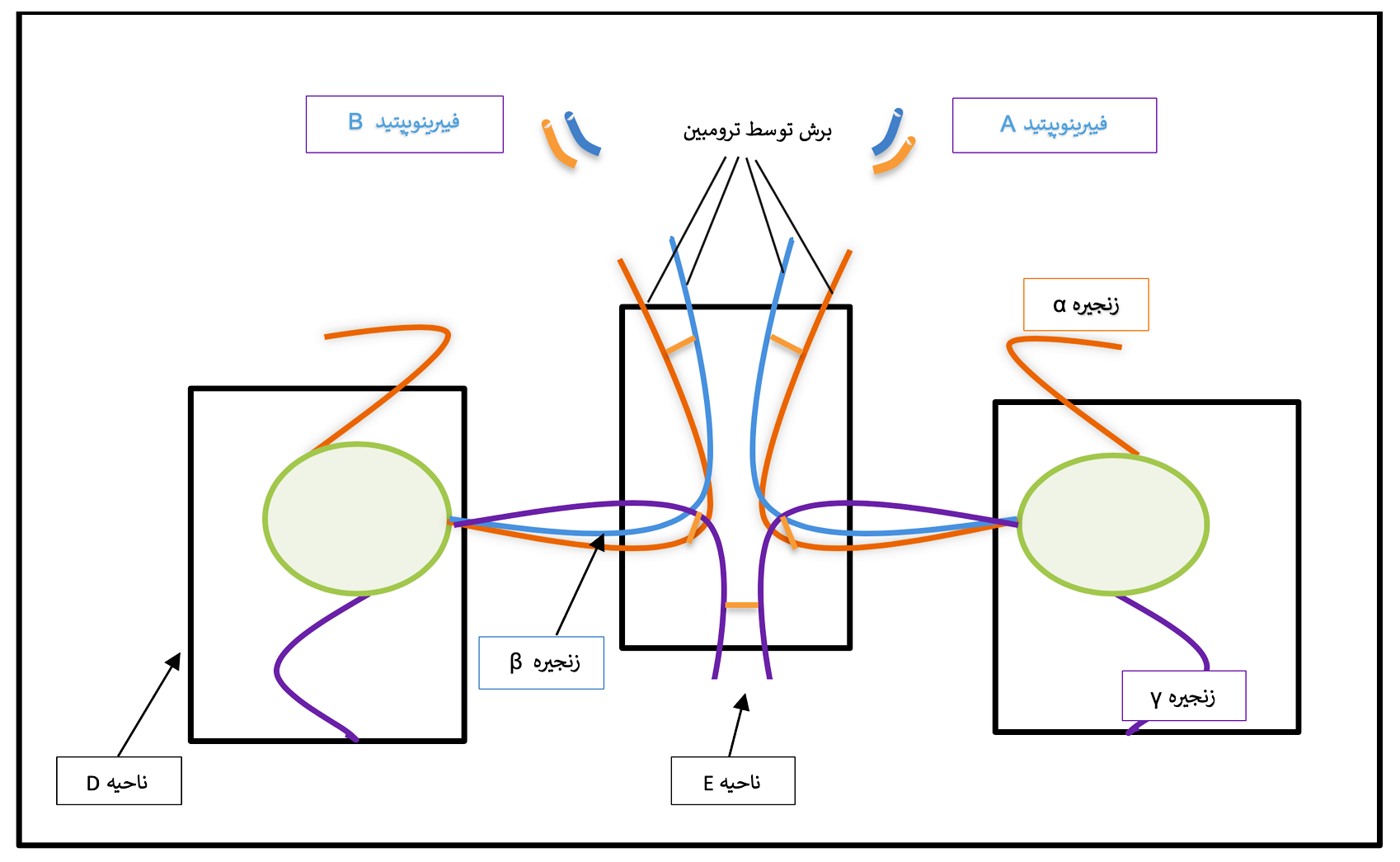
شکل ۱-۶ ساختار ملکول فیبرینوژن
فیبرینوژن از سه زنجیره پلیپپتیدی Aα، Bβ و γ تشکیل شده است. این زنجیرهها یک ساختار دایمر متقارن که با پیوندهای دیسولفیدی به هم متصل هستند را تشکیل میدهند. انتهای آمینی سه زنجیره، ناحیه مرکزی E را شکل میدهد. ناحیه D شامل باقیمانده پایانه کربوکسیل زنجیرههای Bβ و γ است. طی تبدیل فیبرینوژن به فیبرین، فیبرینوپپتیدهای A و B توسط ترومبین برش خورده و آزاد میشوند
اختلالات ارثی فیبرینوژن
اختلالات ارثی فیبرینوژن، اختلالات انعقادی نادری هستند که میتوانند کمیت (تایپ ۱) (آفیبرینوژنمی[6] و هیپوفیبرینوژنمی[7])، کیفیت (تایپ ۲) (دیسفیبرینوژنمی)[8] و یا هر دو جنبه (هیپودیسفیبرینوژنمی)[9] ملکول فیبرینوژن را تحت تأثیر قرار دهند. شیوع دقیق این اختلالات به دلیل وجود تعداد قابل توجه بیماران بدون علامت بالینی، مشخص نیست. آفیبرینوژنمی با الگوی توارث اتوزومال مغلوب به ارث میرسد و هیپوفیبرینوژنمی و دیسفیبرینوژنمی با الگوی توارث اتوزومال غالب به ارث میرسند (جدول ۱-۶) (43, 42, 3). این اختلالات علاوه بر طبقهبندی بر اساس زیرگروهها، بر اساس سطح فیبرینوژن به سه دسته خفیف (g/L >1)، متوسط (g/L0/5-1) و شدید (غیرقابل اندازهگیری) تقسیمبندی میشوند (45, 44). علائم بالینی بیماران به سطح فیبرینوژن و زیرگروه بیماری بستگی دارد (جدول ۱-۶).
اختلالات مادرزادی فیبرینوژن، ۸ درصد اختلالات خونریزی دهنده نادر (RBDs)[10] را در برگرفته و در بین RBDها سومین اختلال شایع هستند (2). تشخیص این بیماری، بر اساس علائم بالینی و ارزیابی مناسب آزمایشگاهی صورت می گیرد. با این حال، بعضی مواقع حتی در آزمایشگاههای پیشرفته، خصوصاً در موارد دیسفیبرینوژنمی و هیپودیسفیبرینوژنمی، روند تشخیص میتواند چالشبرانگیز باشد (جدول ۱-۶) (48-46).
| جدول ۱-۶ طبقهبندی اختلالات مادرزادی فیبرینوژن | ||||
| هیپودیسفیبرینوژنمی | دیسفیبرینوژنمی | هیپوفیبرینوژنمی | آفیبرینوژنمی | |
| نامشخص | نامشخص | نامشخص | 1 در 1 میلیون | شیوع |
| عمدتاً اتوزومال غالب | عمدتاً اتوزومال غالب | عمدتاً اتوزومال غالب | اتوزومال مغلوب | وراثت |
| <1/5 g/L | <1/5 g/L | <1/5 g/L | غیرقابل اندازهگیری | سطح فعالیت |
| <1/5 g/L | >1/5 g/L | <1/5 g/L | غیرقابل اندازهگیری | سطح آنتیژن |
|
– ترومبوز – خونریزی -سقط – خونریزی پس از زایمان |
– عمدتاً فاقد علائم بالینی
– ترومبوز – خونریزی – آمیلوئیدوز – افزایش فشارخون ریوی – سقط -خونریزی پس از زایمان |
– عمدتاً فاقد علائم بالینی (خونریزی بر اساس سطح فیبرینوژن رخ میدهد)
– بیماری ذخیرهای فیبرینوژن – جدا شدن جفت – سقط – خونریزی پس از زایمان |
– خونریزی
– ترومبوز -کیست استخوانی – پارگی خودبخودی طحال – بهبودی ناقص زخم – سقط – خونریزی پس از زایمان |
علائم بالینی |
منابع:
- Neerman-Arbez M, De Moerloose P, Casini A, editors. Laboratory and genetic investigation of mutations accounting for congenital fibrinogen disorders. Seminars in thrombosis and hemostasis; 2016: Thieme Medical Publishers.
- Palla R, Peyvandi F, Shapiro ADJB. Rare bleeding disorders: diagnosis and treatment. 2015;125(13):2052-61.
- Casini A, De Moerloose P, Neerman-Arbez M, editors. Clinical features and management of congenital fibrinogen deficiencies. Seminars in thrombosis and hemostasis; 2016: Thieme Medical Publishers.
- Dorgalaleh A, Alavi SER, Tabibian S, Soori S, Moradi Eh, Bamedi T, et al. Diagnosis, clinical manifestations and management of rare bleeding disorders in Iran. 2017;22(4):224-30.
- De Moerloose P, Casini A, Neerman-Arbez M, editors. Congenital fibrinogen disorders: an update. Seminars in thrombosis and hemostasis; 2013: Thieme Medical Publishers.
- Menache DJAotNYAoS. Congenital fibrinogen abnormalities. 1983;408(1):121-30.
- Casini A, Neerman‐Arbez M, Ariens R, De Moerloose PJJoT, Haemostasis. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. 2015;13(6):909-19.
- De Moerloose P, Neerman-Arbez MJEoobt. Treatment of congenital fibrinogen disorders. 2008;8(7):979-92.
- Molmenti EP, Ziambaras T, Perlmutter DJJoBC. Evidence for an acute phase response in human intestinal epithelial cells. 1993;268(19):14116-24.
- Lee SY, Lee KP, Lim JWJT, haemostasis. Identification and biosynthesis of fibrinogen in human uterine cervix carcinoma cells. 1996;75(03):466-70.
- Haidaris PJSJB. Induction of fibrinogen biosynthesis and secretion from cultured pulmonary epithelial cells. 1997;89(3):873-82.
- Soria J, Soria C, Samama M, Poirot E, Kling CJP-b. Human platelet fibrinogen: a protein different from plasma fibrinogen. 1976;24:15-7.
- Francis CW, Nachman RL, Marder VJJT, haemostasis. Plasma and platelet fibrinogen differ in γ chain content. 1984;52(01):084-8.
- Söderqvist T, Blombäck BJN. Fibrinogen structure and evolution. 1971;58(1):16-23.
- Blombäck BJTr. Fibrinogen structure, activation, polymerization and fibrin gel structure. 1994;75(3):327.
- Mosesson MW, editor Fibrinogen structure and fibrin clot assembly. Seminars in thrombosis and hemostasis; 1998: Copyright© 1998 by Thieme Medical Publishers, Inc.
- Fish RJ, Neerman-Arbez MJT, haemostasis. Fibrinogen gene regulation. 2012;108(09):419-26.
- Henry I, Uzan G, Weil D, Nicolas H, Kaplan J, Marguerie C, et al. The genes coding for A alpha-, B beta-, and gamma-chains of fibrinogen map to 4q2. 1984;36(4):760.
- Crabtree GR, Comeau CM, Fowlkes DM, Fornace Jr AJ, Malley JD, Kant JAJJomb. Evolution and structure of the fibrinogen genes: random insertion of introns or selective loss? 1985;185(1):1-19.
- Asselta R, Duga S, Tenchini MLJJoT, Haemostasis. The molecular basis of quantitative fibrinogen disorders. 2006;4(10):2115-29.
- Macrae FL, Domingues MM, Casini A, Ariens RA, editors. The (Patho) physiology of Fibrinogen γ′. Seminars in thrombosis and hemostasis; 2016: Thieme Medical Publishers.
- De Moerloose P, Neerman-Arbez M, editors. Congenital fibrinogen disorders. Seminars in thrombosis and hemostasis; 2009: © Thieme Medical Publishers.
- Neerman-Arbez M, Tirefort Y, de Moerloose PJJOCD. Can mutations identified in congenital fibrinogen disorders explain the clinical manifestations? 2010;2(2).
- Stanciakova L, Kubisz P, Dobrotova M, Stasko JJEroh. Congenital afibrinogenemia: From etiopathogenesis to challenging clinical management. 2016;9(7):639-48.
- Ogata Y, Hepplmann CJ, Charlesworth MC, Madden BJ, Miller MN, Kalli KR, et al. Elevated levels of phosphorylated fibrinogen-α-isoforms and differential expression of other post-translationally modified proteins in the plasma of ovarian cancer patients. 2006;5(12):3318-25.
- Parastatidis I, Thomson L, Burke A, Chernysh I, Nagaswami C, Visser J, et al. Fibrinogen β-chain tyrosine nitration is a prothrombotic risk factor. 2008;283(49):33846-53.
- Medved L, Weisel J, FIBRINOGEN, THROMBOSIS FXSOTSSCOTISO, Thrombosis HJJo, Haemostasis. Recommendations for nomenclature on fibrinogen and fibrin. 2009;7(2):355-9.
- Mosesson MJJoT, Haemostasis. Fibrinogen and fibrin structure and functions. 2005;3(8):1894-904.
- Olexa SA, Budzynski AZJPotNAoS. Evidence for four different polymerization sites involved in human fibrin formation. 1980;77(3):1374-8.
- Gorkun OV, Veklich YI, Medved’ LV, Henschen AH, Weisel JWJB. Role of the. alpha. C Domains of Fibrin in Clot Formation. 1994;33(22):6986-97.
- Li X, Galanakis D, Gabriel DAJJoBC. Transient intermediates in the thrombin activation of fibrinogen evidence for only the desAA species. 1996;271(20):11767-71.
- Dorgalaleh A, Rashidpanah J. Blood coagulation factor XIII and factor XIII defciency. Blood Rev. 2016;30(6):461–75.
- Muszbek L, Adany R, Mikkola HJCricls. Novel aspects of blood coagulation factor XIII. I. Structure, distribution, activation, and function. 1996;33(5):357-421.
- Muszbek L, Yee VC, Hevessy ZJTR. Blood coagulation factor XIII: structure and function. 1999;94(5):271-305.
- Dorgalaleh A, Rashidpanah JJBr. Blood coagulation factor XIII and factor XIII deficiency. 2016;30(6):461-75.
- Drew AF, Liu H, Davidson JM, Daugherty CC, Degen JL. Wound-healing defects in mice lacking fbrinogen. Blood. 2001;97(12):3691–8.
- Clark RA. Fibrin and wound healing. Ann N Y Acad Sci. 2001;936(1):355–67.
- Ikeda Y, Handa M, Kawano K, Kamata T, Murata M, Araki Y, et al. The role of von Willebrand factor and fbrinogen in platelet aggregation under varying shear stress. J Clin Investig. 1991;87(4):1234.
- Staton CA, Brown NJ, Lewis CE. The role of fbrinogen and related fragments in tumour angiogenesis and metastasis. Expert Opin Biol Ther. 2003;3(7):1105–20.
- Rybarczyk BJ, Lawrence SO, Simpson-Haidaris PJ. Matrix-fbrinogen enhances wound closure by increasing both cell proliferation and migration. Blood. 2003;102(12):4035–43.
- Suh TT, Holmbäck K, Jensen NJ, Daugherty CC, Small K, Simon DI, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fbrinogen-defcient mice. Genes Dev. 1995;9(16):2020–33.
- Cheresh DA, Berliner SA, Vicente V, Ruggeri ZM. Recognition of distinct adhesive sites on fbrinogen by related integrins on platelets and endothelial cells. Cell. 1989;58(5):945–53.
- Peyvandi F. Epidemiology and treatment of congenital fbrinogen defciency. Thromb Res. 2012;130:S7–S11.
43. El Boussaadni Y, Benajiba N, El Ouali A, Amrani R, Rkain M. Congenital afbrinogenemia: a case report. Archives de pediatrie: organe offciel de la Societe francaise de. Pediatrie. 2015;22(1):50–2. - Peyvandi F, Palla R, Menegatti M, Siboni S, Halimeh S, Faeser B, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012;10(4):615–21.
45. Peyvandi F, Di Michele D, Bolton-Maggs P, Lee C, Tripodi A, Srivastava A. Classifcation of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity. J Thromb Haemost. 2012;10(9):1938–43.
46. Lebreton A, Casini A. Diagnosis of congenital fbrinogen disorders. Ann Biol Clin. 2016;74:405–12.
47. Cunningham MT, Brandt JT, Laposata M, Olson JD. Laboratory diagnosis of dysfbrinogenemia. Arch Pathol Lab Med. 2002;126(4):499–505.
48. Miesbach W, Schenk J, Alesci S, Lindhoff-Last E. Comparison of the fbrinogen Clauss assay and the fbrinogen PT derived method in patients with dysfbrinogenemia. Thromb Res. 2010;126(6):e428–e33. - Al-Mondhiry H, Ehmann WC. Congenital afbrinogenemia. Am J Hematol. 1994;46(4):343–7.
- Henselmans J, Meijer K, Haaxma R, Hew J, van Der Meer J. Recurrent spontaneous intracerebral hemorrhage in a congenitally afbrinogenemic patient. Stroke. 1999;30(11):2479–82.
- Taslimi R, Golshani K. Thrombotic and hemorrhagic presentation of congenital hypo/afbrinogenemia. Am J Emerg Med. 2011;29(5):573.e3–5.
52. Paolini R, Sartori MT, Fiorin F, Gorinati M, Boeri G, Girolami A. Perinatal intracranial hemorrhage as frst manifestation of congenital hypofbrinogenemia. Clin Appl Thromb Hemost. 1996;2(1):60–3. - Fettah A, Gökçebay DG, Çulha V, Yaralı N, Tunç B, Özbek N. A rare complication of congenital afbrinogenemia: bone cysts. Turk J Haematol. 2017;34(2):183. 54. Ehmann WC, Al-Mondhiry H. Congenital afbrinogenemia and splenic rupture. Am J Med. 1994;96(1):92–4.
- Dorgalaleh A, Naderi M, Shamsizadeh M. Morbidity and mortality in a large number of Iranian patients with severe congenital factor XIII defciency. Ann Hematol. 2016;95(3):451–5.
- Trehan A, Fergusson I. Congenital afbrinogenaemia and successful pregnancy outcome. Case report. Br J Obstet Gynaecol. 1991;98(7):722–4.
57. Zdziarska J, Undas A, Basa J, Iwaniec T, Skotnicki AB, De Moerloose P, et al. Severe bleeding and miscarriages in a hypofbrinogenemic woman heterozygous for the γAla82Gly mutation. Blood Coagul Fibrinolysis. 2009;20(5):374–6. - Santoro C, Massaro F, Venosi S, Capria S, Baldacci E, Foà R, Mazzucconi MG. Severe thrombotic complications in congenital afbrinogenemia: a pathophysiological and management dilemma. Semin Thromb Hemost. 2016;42(5):577–82.
- Rottenstreich A, Lask A, Schliamser L, Zivelin A, Seligsohn U, Kalish Y. Thromboembolic events in patients with severe inherited fbrinogen defciency. J Thromb Thrombolysis. 2016;42(2):261–6.
- Sartori MT, Milan M, de Bon E, Fadin M, Pesavento R, Zanon E. Thrombosis of abdominal aorta in congenital afbrinogenemia: case report and review of literature. Haemophilia. 2015;21(1):88–94.
- Frenkel E, Duksin C, Herman A, Sherman DJ. Congenital hypofbrinogenemia in pregnancy: report of two cases and review of the literature. Obstet Gynecol Surv. 2004;59(11):775–9.
- Awasthy N, Aggarwal K, Gupta H, Saluja S. Congenital hypofbrinogenemia. Indian Pediatr. 2004;41(2):185–6.
63. Hasselback R, Marion RB, Thomas J. Congenital hypofbrinogenemia in fve members of a family. Can Med Assoc J. 1963;88(1):19.
64. Bay A, Coskun E, Leblebisatan G, Sivasli E. Epidural hematoma and cephalohematoma with congenital hypofbrinogenemia. Blood Coagul Fibrinolysis. 2012;23(3):229–31. - Rubbia-Brandt L, Neerman-Arbez M, Rougemont A-L, Malé P-J, Spahr L. Fibrinogen gamma375 arg→ trp mutation (fbrinogen aguadilla) causes hereditary hypofbrinogenemia, hepatic endoplasmic reticulum storage disease and cirrhosis. Am J Surg Pathol. 2006;30(7):906–11.
- Wehinger H, Klinge O, Alexandrakis E, Schürmann J, Witt J, Seydewitz H. Hereditary hypofbrinogenemia with fbrinogen storage in the liver. Eur J Pediatr. 1983;141(2):109–12.
- Pfeifer U, Ormanns W, Klinge O. Hepatocellular fbrinogen storage in familial hypofbrinogenemia. Virchows Arch B. 1981;36(1):247–55.
68. Casini A, Sokollik C, Lukowski S, Lurz E, Rieubland C, Moerloose P, et al. Hypofbrinogenemia and liver disease: a new case of Aguadilla fbrinogen and review of the literature. Haemophilia. 2015;21(6):820–7. - Asselta R, Robusto M, Braidotti P, Peyvandi F, Nastasio S, D’antiga L, et al. Hepatic fbrinogen storage disease: identifcation of two novel mutations (p. Asp316Asn, fbrinogen Pisa and p. Gly366Ser, fbrinogen Beograd) impacting on the fbrinogen γ-module. J Thromb Haemost. 2015;13(8):1459–67.
- Puls F, Goldschmidt I, Bantel H, Agne C, Bröcker V, Dämmrich M, et al. Autophagyenhancing drug carbamazepine diminishes hepatocellular death in fbrinogen storage disease. J Hepatol. 2013;59(3):626–30.
- Casini A, Duval C, Pan X, Tintillier V, Biron-Andreani C, Ariëns R. Fibrin clot structure in patients with congenital dysfbrinogenaemia. Thromb Res. 2016;137:189–95.
- Casini A, Blondon M, Lebreton A, Koegel J, Tintillier V, de Maistre E, et al. Natural history of patients with congenital dysfbrinogenemia. Blood. 2015;125(3):553–61.
- Miesbach W, Galanakis D, Scharrer I. Treatment of patients with dysfbrinogenemia and a history of abortions during pregnancy. Blood Coagul Fibrinolysis. 2009;20(5):366–70.
- Miesbach W, Scharrer I, Henschen A, Neerman-Arbez M, Spitzer S, Galanakis D. Inherited dysfbrinogenemia: clinical phenotypes associated with fve different fbrinogen structure defects. Blood Coagul Fibrinolysis. 2010;21(1):35–40.
- Hayes T. Dysfbrinogenemia and thrombosis. Arch Pathol Lab Med. 2002;126(11):1387–90.
- Rowczenio D, Stensland M, de Souza GA, Strøm EH, Gilbertson JA, Taylor G, et al. Renal amyloidosis associated with fve novel variants in the fbrinogen A alpha chain protein. Kidney Int Rep. 2017;2:461–9.
- Casini A, Brungs T, Lavenu-Bombled C, Vilar R, Neerman-Arbez M, Moerloose P. Genetics, diagnosis and clinical features of congenital hypodysfbrinogenemia: a systematic literature review and report of a novel mutation. J Thromb Haemost. 2017;15(5):876–88.
- Deering SH, Landy HJ, Tchabo N, Kessler C. Hypodysfbrinogenemia during pregnancy, labor, and delivery. Obstet Gynecol. 2003;101(5, Part 2):1092–4.
- Cheah CY, Brennan SO, Kennedy H, Januszewicz EH, Maxwell E, Burbury K. Fibrinogen Melbourne: a novel congenital hypodysfbrinogenemia caused by γ326Cys-Phe in the fbrinogen γ chain, presenting as massive splanchnic venous thrombosis. Blood Coagul Fibrinolysis. 2012;23(6):563–5.
- Ebert RF, Bell WR. Fibrinogen Baltimore II: congenital hypodysfbrinogenemia with delayed release of fbrinopeptide B and decreased rate of fbrinogen synthesis. Proc Natl Acad Sci. 1983;80(23):7318–22.
- Lefebvre P, Velasco PT, Dear A, Lounes KC, Lord ST, Brennan SO, et al. Severe hypodysfbrinogenemia in compound eterozygotes of the fbrinogen AαIVS4+ 1G> T mutation and an AαGln328 truncation (fbrinogen Keokuk). Blood. 2004;103(7):2571–6.
82. Jayo A, Arnold E, González-Manchón C, Green D, Lord ST. Hypodysfbrinogenemia causing mild bleeding and thrombotic complications in a compound heterozygote of AαIVS4+ 1G> T mutation and Aα4841delC truncation (AαPerth). Thromb Haemost. 2009;101(4):770. - Duga S, Asselta R, Santagostino E, Zeinali S, Simonic T, Malcovati M, et al. Missense mutations in the human β fbrinogen gene cause congenital afbrinogenemia by impairing fbrinogen secretion. Blood. 2000;95(4):1336–41. 84. Neerman-Arbez M, De Moerloose P, Bridel C, Honsberger A, Schönbörner A, Rossier C, et al. Mutations in the fbrinogen Aα gene account for the majority of cases of congenital afbrinogenemia. Blood. 2000;96(1):149–52.
- Neerman-Arbez M. The molecular basis of inherited afbrinogenaemia. Thromb Haemost. 2001;86(1):154–63.
86. Uzan G, Courtois G, Besmond C, Frain M, Sala-Trepat J, Kahn A, et al. Analysis of fbrinogen genes in patients with congenital afbrinogenemia. Biochem Biophys Res Commun. 1984;120(2):376–83. - Neerman-Arbez M, De Moerloose P. Mutations in the fbrinogen gene cluster accounting for congenital afbrinogenemia: an update and report of 10 novel mutations. Hum Mutat. 2007;28(6):540.
- Brennan SO, Fellowes AP, George PM. Molecular mechanisms of hypo-and afbrinogenemia. Ann N Y Acad Sci. 2001;936(1):91–100.
89. Neerman-Arbez M, De Moerloose P, Honsberger A, Parlier G, Arnuti B, Biron C, et al. Molecular analysis of the fbrinogen gene cluster in 16 patients with congenital afbrinogenemia: novel truncating mutations in the FGA and FGG genes. Hum Genet. 2001;108(3):237–40. - Neerman-Arbez M, Vu D, Abu-Libdeh B, Bouchardy I, Morris MA. Prenatal diagnosis for congenital afbrinogenemia caused by a novel nonsense mutation in the FGB gene in a Palestinian family. Blood. 2003;101(9):3492–4.
- Asselta R, Spena S, Duga S, Peyvandi F, Malcovati M, Mannucci PM, et al. Analysis of Iranian patients allowed the identifcation of the frst truncating mutation in the fbrinogen Bbeta-chain gene causing afbrinogenemia. Haematologica. 2002;87(8):855–9.
92. Brennan SO, Davis RL, Conard K, Savo A, Furuya KN. Novel fbrinogen mutation γ314Thr→ Pro (fbrinogen AI duPont) associated with hepatic fbrinogen storage disease and hypofbrinogenaemia. Liver Int. 2010;30(10):1541–7. - Lee MJ, Venick R, Bhuta S, Li X, Wang HL. Hepatic fbrinogen storage disease in a patient with hypofbrinogenemia: report of a case with a missense mutation of the FGA gene. Semin Liver Dis. 2015;35:439–43.
- Asselta R, Platè M, Robusto M, Borhany M, Guella I, Soldà G, et al. Clinical and molecular characterisation of 21 patients affected by quantitative fbrinogen defciency. Thromb Haemost. 2015;113(3):567–76.
- Sumitha E, Jayandharan G, Arora N, Abraham A, David S, Devi G, et al. Molecular basis of quantitative fbrinogen disorders in 27 patients from India. Haemophilia. 2013;19(4):611–8.
- Koopman J, Haverkate F, Grimbergen J, Egbring R, Lord S. Fibrinogen Marburg: a homozygous case of dysfbrinogenemia, lacking amino acids A alpha 461-610 (Lys 461 AAA–> stop TAA). Blood. 1992;80(8):1972–9.
- Vu D, Neerman-Arbez M. Molecular mechanisms accounting for fbrinogen defciency: from large deletions to intracellular retention of misfolded proteins. J Thromb Haemost. 2007;5(s1):125–31.
- Zhou J, Ding Q, Chen Y, Ouyang Q, Jiang L, Dai J, et al. Clinical features and molecular basis of 102 Chinese patients with congenital dysfbrinogenemia. Blood Cell Mol Dis. 2015;55(4):308–15.
- Hill M, Dolan G. Diagnosis, clinical features and molecular assessment of the dysfbrinogenaemias. Haemophilia. 2008;14(5):889–97.
100. Verhovsek M, Moffat KA, Hayward CP. Laboratory testing for fbrinogen abnormalities. Am J Hematol. 2008;83(12):928–31.
101. Sadeghian MH, Keramati MR, Badiei Z, Ravarian M, Ayatollahi H, Rafatpanah H, et al. Alloimmunization among transfusion-dependent thalassemia patients. Asian J Transfus Sci. 2009;3(2):95. - Ridgway HJ, Brennan SO, Faed JM, George PM. Fibrinogen Otago: a major α chain truncation associated with severe hypofbrinogenaemia and recurrent miscarriage. Br J Haematol. 1997;98(3):632–9.
- Martinez J, Holburn R, Shapiro S, Erslev A. Fibrinogen Philadelphia. A hereditary hypodysfbrinogenemia characterized by fbrinogen hypercatabolism. J Clin Investig. 1974;53(2):600.
- Mukai S, Nagata K, Ikeda M, Arai S, Sugano M, Honda T, et al. Genetic analyses of novel compound heterozygous hypodysfbrinogenemia, Tsukuba I: FGG c. 1129+ 62_65 del AATA and FGG c. 1299+ 4 del A. Thromb Res. 2016;148:111–7.
105. Harr JN, Moore EE, Ghasabyan A, Chin TL, Sauaia A, Banerjee A, et al. Functional fbrinogen assay indicates that fbrinogen is critical in correcting abnormal clot strength following trauma. Shock. 2013;39(1):45. - Mackie I, Lawrie A, Kitchen S, Gaffney P, Howarth D, Lowe G, et al. A performance evaluation of commercial fbrinogen reference preparations and assays for Clauss and PT-derived fbrinogen. Thromb Haemost. 2002;87(6):997–1005.
- Mackie IJ, Kitchen S, Machin SJ, Lowe G. Guidelines on fbrinogen assays. Br J Haematol. 2003;121(3):396–404.
- Exner T, Burridge J, Power P, Rickard KA. An evaluation of currently available methods for plasma fbrinogen. Am J Clin Pathol. 1979;71(5):521–7.
- Desvignes P, Bonnet P. Direct determination of plasma fbrinogen levels by heat precipitation. A comparison of the technique against thrombin clottable fbrinogen with spectrophotometry and radial immuno-diffusion. Clin Chim Acta. 1981;110(1):9–17.
110. Besser MW, MacDonald SG. Acquired hypofbrinogenemia: current perspectives. J Blood Med. 2016;7:217. - Casini A, de Moerloose P. Can the phenotype of inherited fbrinogen disorders be predicted? Haemophilia. 2016;22(5):667–75.
- Shapiro SE, Phillips E, Manning RA, Morse CV, Murden SL, Laffan MA, et al. Clinical phenotype, laboratory features and genotype of 35 patients with heritable dysfbrinogenaemia. Br J Haematol. 2013;160(2):220–7.
- Godal H, Brosstad F, Kierulf P. Three new cases of an inborn qualitative fbrinogen defect (Fibrinogen Oslo II). Eur J Haematol. 1978;20(1):57–62.
- Acharya S, Coughlin A, Dimichele DM. Rare Bleeding Disorder Registry: defciencies of factors II, V, VII, X, XIII, fbrinogen and dysfbrinogenemias. J Thromb Haemost. 2004;2(2):248–56.
- Casini A, de Moerloose P, Congenital Fibrinogen Disorders Group. Management of congenital quantitative fbrinogen disorders: a Delphi consensus. Haemophilia. 2016;22(6):898–905.
- Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, et al. Guideline for the diagnosis and management of the rare coagulation disorders. Br J Haematol. 2014;167(3):304–26.
- Bolton-Maggs PH, Perry DJ, Chalmers EA, Parapia LA, Wilde JT, Williams MD, Collins PW, Kitchen S, Dolan G, Mumford AD. The rare coagulation disorders – review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004;10(5):593–628.
[1] Congenital Fibrinogen Disorders
[2] Alternative splicing
[3] Posttranslational modifcations
[4] Sialylation
[5] Three-nodular structure
[6] Afibrinogenemia
[7] Hypofibrinogenemia
[8] Dysfibrinogenemia
[9] Hypodysfibrinogenemia
[10] Rare Bleeding Disorders
microRNAها در هموستاز و ترومبوز
استرس اکسیداتیو و سیستم دفاع آنتیاکسیدانی
پاتوفیزیولوژی و یافتههای آزمایشگاهی کواگولوپاتی ناشی از کووید-19
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام