تشخیص آزمایشگاهی و درمان کمبود توأم فاکتورهای انعقادی
کمبود مشترک فاکتور V و فاکتور VIII
اکبر درگلاله1، حسن مروتی2
1: گروه هماتولوژی و طب انتقال خون، دانشگاه علوم پزشکی ایران
2: مرکز تحقیقات واکسن و سرمسازی رازی
پیشگفتار
کمبود مشترک فاکتور V و فاکتور VIII (F5F8D) یک اختلال خونریزیدهنده نادر[1](RBD) است که 3% از کل موارد RBD را شامل میشود و شایعترین شکل از کمبود مرکب فاکتورهای انعقادی (MCFD) خانوادگی است (جدول 1-9). شیوع این اختلال حدود 1 در یک میلیون نفر در جمعیت عمومی است و در بین یهودیان خاورمیانه و ایرانیان غیریهودی بسیار شایعتر است (1 در 100000 نفر) (1, 2). این شیوع بالاتر را شاید بتوان با میزان بالای ازدواجهای فامیلی در این مناطق توجیه کرد (3). علت زمینهای F5F8D متفاوت از کمبود مجزای فاکتور V و فاکتور VIII است که به ترتیب ناشی از نقص ژنی در ژنهای F5 و F8 است. F5F8D یک اختلال اتوزوم مغلوب است که ناشی از موتاسیون در ژنهای لکتین متصلشونده به مانوز-1[2](LMAN1) یا کمبود مرکب فاکتور انعقادی-2[3](MCFD2) است (4). F5F8D معمولاً با تمایل خفیف به خونریزی، عمدتاً خونریزیهای پوستی مخاطی، همراه است؛ بنابراین معمولاً درمان تقاضامحور[4] برای مدیریت بیماری کافی است و بهطور کلی برای بیماران با این اختلال نیازی به پروفیلاکسی نیست (5). تشخیص بیماری عمدتاً بر اساس طولانی شدن زمان پروترومبین [5](PT) و زمان ترومبوپلاستین نسبی فعالشده [6](aPTT) و کاهش همزمان فعالیت فاکتورهای V و VIII است؛ اما لازم است که از وجود همزمان کمبود فاکتور V با هموفیلی A یا بیماری فونویلبراند [7](VWD)، افتراق داده شود.
| جدول 1-9 خصوصیات اصلی کمبود مرکب فاکتورهای انعقادی (MCFD) | |||||||
| کمبود فاکتور | عامل زمینه | نامگذاری OMIM | |||||
| اکتسابی | |||||||
| بیماری کبدی | کمبود جهانی متغیر | سنتز معیوب | – | ||||
| DIC | کمبود جهانی متغیر | مصرف بیش از حد | – | ||||
| انتقال خون حجیم | کمبود جهانی متغیر | اثر ترقیقی | – | ||||
| بایپس قلبی-ریوی | کمبود جهانی متغیر | اثر ترقیقی | – | ||||
| خانوادگی (MCFD) | |||||||
| نقص ژنتیکی منفرد | |||||||
| نقص در یک ژن درگیر در تولید یا فعالسازی چندین فاکتور | |||||||
| F5F8D |
FV + FVIII
|
موتاسیونهای ژن LMAN1 یا MCFD2 | 227300 | ||||
| کمبود مشترک فاکتورهای وابسته به ویتامین K | FII+FVII+FIX+FX | موتاسیونهای ژن GGCX یا VKORC1 |
2777450 607473 |
||||
| FMCFD به عنوان بخشی از یک سندرم | |||||||
| اختلالات مادرزادی گلیکوزیلاسیون | متنوع | موتاسیونهای PMM2 | 212065 (CDG1a) | ||||
| سندرم نونان | متنوع | موتاسیونهای PTPN11، SOS1، RAF1، KRAS | 163950 | ||||
| مشکلات ذاتی متابولیسم کبد | متنوع | موتاسیونهای GALT | 230400 (گالاکتوزمی) | ||||
| سندرم حذف 34q13 | FVII + FX
|
حذف ژنی 34q13 |
227500 227600 |
||||
| دو یا چند نقص مجزا | |||||||
|
OMIM: Online Mendelian Inheritance in Man. (http://www.ncbi.nlm.nih.gov/omim/) DIC: انعقاد داخل عروقی منتشر، FMCFD: کمبود مرکب فاکتور انعقادی خانوادگی، F5F8D: کمبود مشترک فاکتور V و فاکتور VIII، LMAN1: لکتین متصلشونده به مانوز-1، MCFD2: کمبود مرکب فاکتور انعقادی-2، GGCX: γ– گلوتامیل کربوکسیلاز، VKORC1: زیرواحد 1 کمپلکس ویتامین K اپوکسید ردوکتاز، PMM2: فسفومانوموتاز-2، CDGa1: اختلال مادرزادی گلیکوزیلاسیون تایپ 1a، PTPN11: پروتئین تیروزین فسفاتاز غیر رسپتوری تایپ 11، SOS1: son of sevenless homolog 1،GALT : گالاکتوز-1- فسفات یوریدیل ترانسفراز |
|||||||
ساختار و عملکرد LMAN1 و MCFD2
در گذشته LMAN1 به عنوان شاخصی برای جزء واسط 53 کیلودالتونی شبــکه اندوپلاسمی– گلژی [8](ERGIC-53) شناخته میشد. این پروتئین به عنوان یک گیرنده محموله برای گلیکوپروتئینها عمل میکند. نشان داده شده است که LMAN1 یک جزء ضروری در مسیر ترشحی فاکتور V و فاکتور VIII است، بهطوریکه انتقال هر دو فاکتور را از شبکه اندوپلاسمی [9](ER) به گلژی میانجیگری میکند (شکل 1-9) (4، 6, 7).
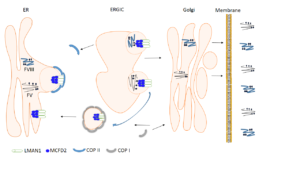
شکل 1-9 انتقال فاکتور V و فاکتور VIII بهواسطه کمپلکس LMAN1 و MCFD2. N– گلیکوزیلاسیون و تشکیل پیوند S-S در فاکتور V و فاکتور VIII در ER انجام میشود. فاکتور V و فاکتور VIII بر روی جایگاههای اتصالی مجزا بـــــه کمپلکس LMAN1-MCFD2 متصل میشوند و در حالــی که داخل COP II بستهبندی میشوند به ERGIC منتقل میگردند. محل دقیق جایگاههای اتصالی مشخص نیست، با این حال به نظر میرسد CRD احتمالاً نقش مهمی در شناسایی فاکتور V و فاکتور VIII دارد. فاکتور V و فاکتور VIII به دستگاه گلژی آزاد میشوند، جایی که سایر اصلاحات پس از ترجمه همچون O– گلیکوزیلاسیون و مراحل بیشتری از N– گلیکوزیلاسیون رخ میدهد. کمپلکس LMAN1 و MCFD2 داخل COP I بستهبندی شده و سپس مجدداً به ER برمیگردند (به عنوان انتقال پسرفتی شناخته میشود). در نهایت، فاکتور V و فاکتور VIII به بیرون از سلول ترشح میشوند. FV: فاکتور V، FVIII: فاکتور VIII، LMAN1: لکتین متصلشونده به مانوز-1، MCFD2: کمبود مرکب فاکتور انعقادی-2، ER: شبکه اندوپلاسمی، ERGIC: جزء واسط گلژی شبکه اندوپلاسمی، COP II: پروتئین پوشاننده II، CRD: دامین شناساییکننده کربوهیدرات، COP I: پروتئین پوشاننده I
LMAN1 یک پروتئین با وزن 53 کیلودالتون، ترانس ممبرن و هموهگزامریک است. LMAN1 دارای سه دامین است، شامل:
(I) یک دامین لومینال که خود به یک دامین شناساییکننده کربوهیدرات[10](CRD) و نواحی فنر فنری شده هلیکس آلفا تقسیم میشود.
(II) یک دامین ترانس ممبرن
(III) یک دامین سیتوپلاسمی (شکل 2-9 ب).
مشخص شده است که یک برهمکنش مستقیم بین LMAN1 و MCFD2 وجود دارد، بهطوریکه هر هگزامر LMAN1 با شش پروتئین MCFD2 مرتبط است (شکل 1-9) (8). MCFD2 یک پروتئین مونومر 16 کیلودالتونی است. این پروتئین دو موتیف متصلشونده به Ca+2 دارد که دامینهای EF-hand نامیده میشوند و توسط یک ناحیه اتصالی[11] از هم جدا شدهاند (شکل 3-9 ب) (4, 7).
کمبود مشترک فاکتور V و فاکتور VIII
F5F8D (OMIM: 227300# و 61362522#) اولین بار توسط Oeri و همکاران در سال 1954 توصیف شد (9). عامل زمینهای F5F8D متفاوت از کمبود منفرد فاکتور V و فاکتور VIII است که به ترتیب ناشی از نقص ژنی، در ژنهای F5 و F8 میباشند. اگرچه اولین مورد F5F8D در سال 1954 گزارش شد، اما اساس مولکولی زمینهساز آن بیش از 40 سال ناشناخته باقی ماند (10). در سال 1998 Nichols و همکارانش موتاسیونهای نول در ژن ERGIC53 (امروزه به عنوان LMAN1 شناخته میشود) را به عنوان نقص ژنتیکی عامل ایجاد F5F8D معرفی کردند (5, 11, 12). 5 سال بعد، بررسی حدود 15% از خانوادههای مبتلا که هیچ موتاسیونی در ERGIC-53 نداشتند نشان داد نقص ژنتیکی دیگری به عنوان عامل بیماری وجود دارد (4).
تظاهرات بالینی
بیماران مبتلا به F5F8D معمولاً رخدادهای خونریزیدهنده شدیدی را نشان نمیدهند. این بیماران عمدتاً کبودیهای آسان، خونریزی از بینی، خونریزی لثه و خونریزی بیش از حد پس از تروما یا اعمال تهاجمی همچون کشیدن دندان را تجربه میکنند. زنان مبتلا معمولاً دچار منوراژی و خونریزیهای پس از زایمان میشوند. همارتروز و خونریزی از دستگاه گوارش نیز ممکن است در این بیماران رخ دهد، اما با وقوع بسیار کمتر. تنها در موارد اندکی هماچوری، هماتوم عضلات و خونریزیهای سیستم اعصاب مرکزی [12](CNS) گزارش شده است (2, 3, 13-15).
بهطور کلی بیماران مبتلا به F5F8D تظاهرات بالینی مختلفی را بروز میدهند که از دورههای خفیف خونریزی همچون خونریزی از بینی تا خونریزیهای شدید از جمله خونریزی CNS متغیر است (جدول 2-9). F5F8Dدر مقایسه با کمبود مجزای فاکتور V و فاکتور VIII، با تمایل بیشتر به خونریزی همراه نیست (5).
| جدول 2-9 تظاهرات بالینی بیماران مبتلا به کمبود مشترک فاکتور V و فاکتور VIII (F5F8D) | |||||
| تظاهرات بالینی | وقوع (تعداد بیماران دارای علامت/ تعداد کلی بیماران) | ||||
| Seligsohn و همکاران
(14=n) |
Peyvandi
و همکاران (27=n) |
Shetty و همکاران(9=n) |
Mansouritorghabeh و همکاران
(19=n) |
Viswabandya و همکاران (37=n) |
|
| خونریزی بینی | 57% | 77/8% | – | 69/2% | 18/9% |
| خونریزی لثه | 64/3% | – | 44/4% | – | 48/6% |
| اکیموز/ کبودی آسان | 28/6% | – | 44/4% | – | 29/7% |
| منوراژی | 100% | 58/3% | 50% | 33/3% | 66/7% |
| خونریزی پس از ختنه* | – | 66/7% | – | 46/1% | – |
| خونریزی شدید پس از کشیدن دندان* | 92/3% | 82/3% | 3/33% | 92/3% | 56/7% |
| خونریزی شدید پس از جراحی* | 75% | 75% | – | 83/3% | 62/2% # |
| خونریزی شدید پس از زایمان* | 100% | 75% | – | 50% | – |
| خونریزی پس از انزال* | 42/8% | – | – | – | – |
| خونریزی پس از سقط جنین* | 80% | – | – | – | – |
| خونریزی پس از بریدگی* | – | – | 77/8% | 57/9% | – |
| همارتروز | – | 26% | – | 36/8% | 13/5% |
| خونریزی از دستگاه گوارش | 21/4% | 7/4% | – | 10/5% | 2/7% |
| هماچوری | 14/3% | – | – | – | – |
| هماتوم عضلات | – | 7/4% | – | – | – |
| خونریزی درون جمجمهای | – | 3/7% | – | – | – |
| * این علامتها در بیمارانی که تحت عمل مربوطه قرار داشتند ارزیابی شده است.
$ یک نوزاد در اثر خونریزی پس از ختنه فوت کرد (در مطالعه مذکور بررسی نشده است). # این تعداد شامل بیمارانی که خونریزی پس از تروما داشتند نیز میشود. |
|||||
اساس مولکولی
به نظر میرسد موتاسیونهای ژنهای LMAN1 و MCFD2 تقریباً مسئول تمامی موارد F5F8D هستند. ژن کدکننده LMAN1 با طولی برابر با 29 کیلوباز و 13 اگزون (شکل 2-9 الف) بر روی بازوی بلند کروموزوم 18 قرار گرفته است (21.32q18). MCFD2 توسط ژنی به طول 19 کیلوباز بر روی بازوی کوتاه کروموزوم 2 (21p2) و شامل 4 اگزون، کد میشود (شکل 3-9 الف).

شکل 2-9 (الف) ژن LMAN1 و موتاسیونهای عامل F5F8D. ژن LMAN1 شامل 13 اگزون است. اگزونها با مستطیلهای خاکستری نشان داده شدهاند و در مقیاس طراحی شدهاند، اینترونها با خطوط زرد نشان داده شدهاند و در مقیاس نیستند. مستطیلهای سفید نشاندهنده ′3 UTR و ′5 UTR ژن هستند. (ب) دامینهای پروتئین LMAN1. سیگنال پپتید LMAN1 در انتقال LMAN1 به ER نقش دارد. این پروتئین از 3 دامین تشکیل شده است: یک دامین لومینال، یک دامین ترانس ممبرن (TM) و یک دامین کوتاه سیتوپلاسمی (C). دامین لومینال به دو زیر دامین تقسیم میشود: یک CRD در انتهای آمینی و یک دامین فنری α– هلیکس پروگزیمال- غشایی که به عنوان دامین stalk شناخته میشود. LMAN1: لکتین متصلشونده به مانوز-1، UTR: ناحیه ترجمهنشده، ER: شبکه اندوپلاسمی، CRD: دامین شناساییکننده کربوهیدرات
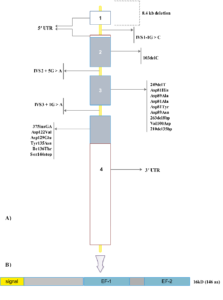
شکل 3-9 (الف) ژن MCFD2 و موتاسیونهای عامل F5F8D. اگزونها با مستطیلهای خاکستری نشان داده شدهاند و در مقیاس طراحی شدهاند. اینترونها با خطوط زرد نشان داده شدهاند و در مقیاس نیستند. مستطیلهای سفیـــــد نشاندهنده 3′UTR و 5′UTR ژن هستند. (ب) دامینهای پروتئین MCFD2. MCFD2 یک پروتئین کوچک با 166 اسید آمینه و 3 دامین است که عبارتند از یک توالی سیگنال برای هدایت MCFD2 به ER و دو موتیف EF-hand که احتمالاً میتوانند به یونهای Ca+2 متصل شوند. MCFD2: کمبود مرکب فاکتور انعقادی-2، UTR: ناحیه ترجمهنشده، ER: شبکه اندوپلاسمی، موتیف EF-hand: موتیف متصلشونده به کلسیم شامل دو هلیکس (E و F) که با یک لوپ به هم وصل شدهاند (هلیکس- لوپ – هلیکس)
تاکنون حداقل 38 موتاسیون در ژن LMAN1 و 20 موتاسیون در ژن MCFD2 توصیف شده است (جدول 3-9 و 4-9) (شکل 2-9 الف و 3-9 الف) (16-22). شایعترین موتاسیونها در ژن LMAN1 موتاسیونهای حذف/اضافه، بیمعنی و اسپلایسسایت هستند که باعث تخریب کامل عملکرد پروتئین میشوند (جدول 3-9). اکثر موتاسیونهایی که ژن MCFD2 را درگیر میکنند موتاسیونهای حذف/اضافه، بد معنی و اسپلایسسایت هستند (جدول 4-9).
| جدول 3-9 موتاسیونهای ژن LMAN1 در بیماران با کمبود مشترک فاکتور V و فاکتور VIII (F5F8D) | ||
| نوع موتاسیون | نامگذاری | ناحیه ژنی |
| کدون آغازی | Met1Thr | اگزون 1 |
| تغییر چهارچوب | 23delG | اگزون 1 |
| تغییر چهارچوب | 31delG | اگزون 1 |
| تغییر چهارچوب | 89insG | اگزون 1 |
| بد معنی | Trp67Ser | اگزون 1 |
| بیمعنی | Gly114stop | اگزون 2 |
| تغییر چهارچوب | 422delC | اگزون 3 |
| تغییر چهارچوب | IVS4+17 del T | اگزون 4 |
| بیمعنی | Arg202stop | اگزون 5 |
| اسپلایسینگ | IVS5 + 1G > T | اینترون 5 |
| تغییر چهارچوب | 720del16bp | اگزون 6 |
| تغییر چهارچوب | 781delT | اگزون 7 |
| تغییر چهارچوب | 795delC | اگزون 7 |
| تغییر چهارچوب | 813del72bp | اگزون 7 |
| اسپلایسینگ | 822G>A | اگزون 7 |
| اسپلایسینگ | IVS7+1G > A | اینترون 7 |
| اسپلایسینگ | IVS7-1G > A | اینترون 7 |
| اسپلایسینگ | IVS 7–1G > C | اینترون 7 |
| اسپلایسینگ | IVS7 + 33insGGTT | اینترون 7 |
| بیمعنی | Lys302stop | اگزون 8 |
| بیمعنی | Gln317stop | اگزون 8 |
| تغییر چهارچوب | 841delA | اگزون 8 |
| تغییر چهارچوب | 912insA | اگزون 8 |
| تغییر چهارچوب | 912delA | اگزون 8 |
| بیمعنی | Glu321stop | اگزون 9 |
| تغییر چهارچوب | 1109delTC | اگزون 9 |
| بیمعنی | Gln380stop | اگزون 9 |
| اسپلایسینگ | IVS9 + 2T > G | اینترون 9 |
| اسپلایسینگ | IVS9 + 2T > C | اینترون 9 |
| تغییر چهارچوب | 1208insT | اگزون 10 |
| تغییر چهارچوب | 1214del5bp | اگزون 10 |
| تغییر چهارچوب | 1261insTG | اگزون 11 |
| بیمعنی | Arg456stop | اگزون 11 |
| اسپلایسینگ | 1271delG | اگزون 11 |
| تغییر چهارچوب | 1356delC | اگزون 11 |
| بد معنی | Cys475Arg | اگزون 12 |
| حذف | 1456delGTG | اگزون 12 |
| تغییر چهارچوب | 1524delA | اگزون 13 |
| LMAN1: لکتین متصلشونده به مانوز- 1 | ||
| جدول 4-9 موتاسیونهای ژن MCFD2 در بیماران با کمبود مشترک فاکتور V و فاکتور VIII (F5F8D) | ||
| نوع موتاسیون | نامگذاری | ناحیه ژنی |
| حذف بزرگ | 8.4 kb deletion | پروموتر و اگزون 1 |
| اسپلایسینگ | IVS1–1G > C | اینترون 1 |
| تغییر چهارچوب | 103delC | اگزون 2 |
| اسپلایسینگ | IVS2 + 5G > A | اینترون 2 |
| تغییر چهارچوب | 249delT | اگزون 3 |
| تغییر چهارچوب | 263del8bp | اگزون 3 |
| تغییر چهارچوب | 210del35bp | اگزون 3 |
| بد معنی | Asp81Tyr | اگزون 3 |
| بد معنی | Asp81His | اگزون 3 |
| بد معنی | Asp81Ala | اگزون 3 |
| بد معنی | Asp89Asn | اگزون 3 |
| بد معنی | Asp89Ala | اگزون 3 |
| بد معنی | Val100Asp | اگزون 3 |
| اسپلایسینگ | IVS3 + 1G > A | اینترون 3 |
| تغییر چهارچوب | 375insGA | اگزون 4 |
| بد معنی | Asp122Val | اگزون 4 |
| بد معنی | Asp129Glu | اگزون 4 |
| بد معنی | Tyr135Asn | اگزون 4 |
| بد معنی | Ile136Thr | اگزون 4 |
| بیمعنی | Ser144stop | اگزون 4 |
| MCFD2: کمبود مرکب فاکتور انعقادی-2 | ||
برخی موتاسیونها همچون 89insG (تغییر چهارچوب)، IVS9+2 T>C (اسپلایسینگ) و M1T (c.2T > C) (متوقفکننده کدون آغازی) که به ترتیب منحصراً در یهودیان خاورمیانه، یهودیان تونسی و تبار ایتالیایی دیده میشوند، بیانگر اثر بنیادین[13] هستند (21, 22).
بر اساس مطالعهای توسط Zhang و همکاران، پیشنهاد شده است که موتاسیونهایی که MCFD2 را درگیر میکنند، در مقایسه با موتاسیونهای LMAN1، با سطوح پایینتر فاکتور V و فاکتور VIII مرتبط هستند. این یافته، یک ارتباط ژنوتیپ- فنوتیپ را در بیماران با F5F8D تأیید میکند (23). در F5F8D، ارتباط ضعیفی بین فعالیت فاکتور V و فاکتور VIII و شدت فنوتیپ بالینی وجود دارد. هتروزیگوتهای F5F8D معمولاً بدون علامت هستند (5).
منابع:
- Castaman G, Linari S. Diagnosis and treatment of von Willebrand disease and rare bleeding disorders. Journal of clinical medicine. 2017;6(4):45.
- Seligsohn U, Zivelin A, Zwang E. Combined factor V and factor VIII deficiency among non-Ashkenazi Jews. New England Journal of Medicine. 1982;307(19):1191-5.
- Peyvandi F, Tuddenham E, Akhtari A, Lak M, Mannucci P. Bleeding symptoms in 27 Iranian patients with the combined deficiency of factor V and factor VIII. British journal of haematology. 1998;100(4):773-6.
- Zhang B. Recent developments in the understanding of the combined deficiency of FV and FVIII. British journal of haematology. 2009;145(1):15-23.
- Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, et al. Guideline for the diagnosis and management of the rare coagulation disorders: a United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British Committee for Standards in Haematology. British journal of haematology. 2014;167(3):304-26.
- Zhang B, Ginsburg D. Familial multiple coagulation factor deficiencies: new biologic insight from rare genetic bleeding disorders. Journal of Thrombosis and Haemostasis. 2004;2(9):1564-72.
- Zheng C, Liu H-h, Yuan S, Zhou J, Zhang B. Molecular basis of LMAN1 in coordinating LMAN1-MCFD2 cargo receptor formation and ER-to-Golgi transport of FV/FVIII. Blood. 2010;116(25):5698-706.
- Zheng C, Liu H-h, Zhou J, Zhang B. EF-hand domains of MCFD2 mediate interactions with both LMAN1 and coagulation factor V or VIII. Blood. 2010;115(5):1081-7.
- Oeri J, Matter M, Isenschmid H, Hauser F, Koller F. Congenital factor V deficiency (parahemophilia) with true hemophilia in two brothers. Bibliotheca paediatrica. 1954;58:575.
- Jin D-Y, Ingram BO, Stafford DW, Tie J-K. Molecular basis of the first reported clinical case of congenital combined deficiency of coagulation factors. Blood. 2017;130(7):948-51.
- Nichols WC, Seligsohn U, Zivelin A, Terry VH, Arnold ND, Siemieniak DR, et al. Linkage of combined factors V and VIII deficiency to chromosome 18q by homozygosity mapping. The Journal of clinical investigation. 1997;99(4):596-601.
- Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, et al. Mutations in the ER–Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93(1):61-70.
- Shetty S, Madkaikar M, Nair S, Pawar A, Baindur S, Pathare A, et al. Combined factor V and VIII deficiency in Indian population. Haemophilia: the official journal of the World Federation of Hemophilia. 2000;6(5):504-7.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA: a cancer journal for clinicians. 2017;67(1):7-30.
- Viswabandya A, Baidya S, Nair S, Lakshmi K, Mathews V, George B, et al. Clinical manifestations of combined factor V and VIII deficiency: a series of 37 cases from a single center in India. American journal of hematology. 2010;85(7):538-9.
- Elmahmoudi H, Wigren E, Laatiri A, Jlizi A, Elgaaied A, Gouider E, et al. Analysis of newly detected mutations in the MCFD2 gene giving rise to combined deficiency of coagulation factors V and VIII. Haemophilia. 2011;17(5):e923-e7.
- Hejer E, Adnen L, Asma J, Ibtihel M, Benammar-Elgaaied A, Gouider E. Identification of a novel mutation in the MCFD2 gene in a Tunisian family with combined factor V and VIII deficiency. La Tunisie medicale. 2012;90(4):343-4.
- Karimi M, Cairo A, Safarpour MM, Haghpanah S, Ekramzadeh M, Afrasiabi A, et al. Genotype and phenotype report on patients with combined deficiency of factor V and factor VIII in Iran. Blood coagulation & fibrinolysis. 2014;25(4):360-3.
- Wang A, Duan Q, Ding K, Liu X, Wu J, Sun Z. Successful abdominal operation without replacement therapy in a patient with combined factor V (FV) and FVIII deficiency due to novel homozygous mutation in LMAN1. Haemophilia. 2015;21(6):e492-e4.
- Wang A, Liu X, Wu J, Cai X, Zhu W, Sun Z. Combined FV and FVIII deficiency (F5F8D) in a Chinese family with a novel missense mutation in MCFD 2 gene. Haemophilia. 2014;20(6):e436-e8.
- Zheng C, Zhang B, editors. Combined deficiency of coagulation factors V and VIII: an update. Seminars in thrombosis and hemostasis; 2013: Thieme Medical Publishers.
- Genotypes of patients with combined factor V and VIII deficiency [internet]. 2011. https://c.ymcdn.com/sites/www.isth.org/resource/resmgr/publications/fv_and_viii_mutations-2011.pdf.
- Zhang B, Spreafico M, Zheng C, Yang A, Platzer P, Callaghan MU, et al. Genotype-phenotype correlation in combined deficiency of factor V and factor VIII. Blood. 2008;111(12):5592-600.
[1] Rare bleeding disorder
[2] Lectin mannose binding 1
[3] Multiple coagulation factor deficiency 2
[4] On-demand therapy
[5] Prothrombin time
[6] Activated partial thromboplastin time
[7] Von Willebrand disease
[8] Endoplasmic reticulum Golgi intermediate compartment 53 kDa
[9] Endoplasmic reticulum
[10] Carbohydrate recognition domain
[11] Linker region
[12] Central nervous system
[13] Funder effect
هموستاز ثانویه و آبشار انعقادی
دستورالعمل راهبردی برای تشخیص کمبود فاکتور سیزده در ایران (4)
روشهای آزمایشگاهی و استانداردهای بینالمللی در تشخیص اختلالات انعقادی (بخش اول)
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام