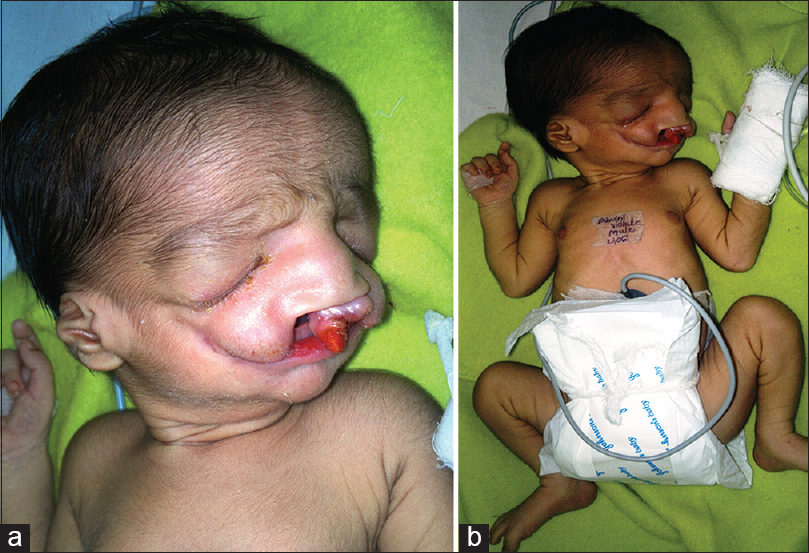
سندرم تریچر کالینز
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، زینب غلامی (دانشجوی هماتولوژی)، دکتر علیاکبر موثقپور اکبری (هماتولوژیست)

سندرم تریچر کالینز یا دیسوستوز ماندیبولوفاشیال یک بیماری نادر اتوزوم غالب است که با بدشکلیهای جمجمهای- صورتی نظیر وجود نداشتن استخوانهای گونه همراه است. سندرم تریچر کالینز ۱ در هر ۵۰۰۰۰ تولد یافت میشود. خصوصیتهای فیزیکی رایج آن عبارتند از: چشمهایی که به پایین نگاه میکنند، میکروگانتیا (فک پایین کوچک)، نداشتن حس شنوایی، زایگومای (استخوان گونه) غیر تکامل یافته، افتادگی بخش جانبی پلکهای پایین و گوشهای بدشکل یا عدم وجود گوش. بعد از اینکه یک جراح و چشم پزشک انگلیسی به نام ادوارد تریچر کالینز آن را در سال ۱۹۰۰ کشف کرد به افتخار او این نام را بر این بیماری نهادند. در سال ۱۹۴۹ آدولف فرانسچتی و دیوید کلین طبق مشاهداتشان نام دیسوستوز ماندیبولوفاشیال را بر آن نهادند که بیشتر خصوصیات بالینی آن را توصیف میکند.

تظاهرات بالینی سندرم تریچر کالینز:
هیپوپلازی فک، اختلالات پلک تحتانی، شکاف پلکی با انحنا به پایین down slanted یا anti-mongoloid و شکاف در قوس زیگوماتیک از شایعترین یافتهها هستند. کلبومای پلک تحتانی معمولاً در یک سوم خارجی پلک دیده میشود و مژههای پلک تحتانی معمولاً در سمت داخل کلبوما یا به صورت کامل وجود ندارند یا ناقص هستند. شکاف در قوس زیگوماتیک سبب ایجاد گونههای فرورفته میشود. فک تحتانی به شدت کوچک است و به راحتی از روی چهره میتوان تشخیص داد ولی گاه نیاز به رادیوگرافی برای تشخیص میباشد. شاخه (ramus) استخوان ماندیبول ممکن است ناقص باشد و زوائد کورونوئید و کوندیلار آن صاف شده یا اصلاً وجود نداشته باشند. اشکالات در ناحیه اطراف گوش و گونهها و شکاف کام نیز از نشانههای دیگر سندرم است.
اختلالات گوش از یافتههای دیگر بیماری است که هر سه قسمت گوش (خارجی، میانی و داخلی) را درگیر میکند. اختلال شنوایی انتقالی در ۲۵ تا ۵۰ درصد بیماران وجود دارد که علت آن عمدتاً هیپوپلازی مجرای گوش خارجی و استخوانچههای گوش میانی میباشد. اختلالات دیگر گوش خارجی به خصوص کوچکی آن (Microtia) شایع است. برجستگی و حفرات پوستی در ناحیه اطراف گوش و گونهها دیده میشوند. شکاف کام در ۲۵ تا ۳۳ درصد بیماران دیده میشود و ممکن است با شکاف لب همراه باشد. افزایش منطقه رویش موها در ناحیه گونهها و بزرگی دهان از سایر یافتهها هستند. به علت فرورفتگی گونهها و فک تحتانی، بینی بیمار بزرگ به نظر میرسد. اختلالات دندانها شامل اختلال اکلوژن و دندانهای جدا از هم، هیپوپلاستیک یا نابجا میباشد.
آترزیکوان هم در برخی بیماران دیده میشود. تغییرات صورت در این بیماران بسیار واضح و اختصاصی است و میتوان با کمی دقت به راحتی این بیماران را تشخیص داد. عقبماندگی ذهنی در ۵٪ بیماران دیده میشود که آن را به هایپوکسی و اختلالات تنفسی در زمان نوزادی نسبت میدهند. بیماریهای مادرزادی قلبی در برخی بیماران دیده میشود و شیوع آن نسبت به افراد طبیعی بیشتر است. بیشترین اختلالات شامل نقص دیواره بطنی(Ventricular Septal Defect) ، نقص دیواره دهلیزی و باز ماندن مجرای شریانی (Patent Ductus Arteriosus) هستند. این بیماران نیز در اکوکاردیوگرافی انجام شده، باز بودن مجرای شریانی و سوراخ بیضی باز را نشان میدهند.

ژنتیک مولکولی سندرم تریچر کالینز:
این بیماری از نظر ژنتیک بیماری به صورت اتوزومال غالب منتقل میشود ولی ۶۰٪ موارد به علت جهش جدید به وجود میآیند. نفوذ ژن بیماری متغیر است و تظاهرات بیماری در یک خانواده متفاوت میباشد. ژن بیماری با نام TCOF1 در سال ۱۹۹۶ شناخته شده و روی کروموزوم شماره 5q32-33.1 قرار دارد.



پیشآگهی در سندرم تریچر کالینز:
مبتلایان به این بیماری برخلاف سایر سندرمهای مادرزادی معمولاً مشکل ذهنی یا اسکلتی ندارند و میتوانند افراد مؤثری برای جامعه باشند. در این میان وجود اختلالات مادرزادی قلبی و تشخیص و درمان به موقع آنها اهمیت افزونتری نسبت به تشخیص و درمان این اختلالات در مبتلایان به سایر سندرمها دارد. گرچه ناهنجاریهای ظاهری سبب بروز مشکلات روحی روانی در سنین مدرسه و بلوغ برای این بیماران میشود، اما مطالعات نشان دادهاند که اکثر این بیماران با یا بدون کمک جراحی ترمیمی با این ناهنجاریها خو میگیرند و در جامعه ادغام میشوند.


درمان سندرم تریچر کالینز:
تا به حال هیچ درمانی برای سندرم تریچر کالینز، یافت نشده است. همانطور که گفته شد افراد مبتلا به سندرم تریچر کالینز، از نظر یادگیری و آموزش هیچگونه مشکلی ندارند و حتی پزشکان و جراحانی مبتلا به سندرم تریچر کالینز هستند.

تنها مسیر درمانی جراحیهای پلاستیک بر پوست صورت افراد مبتلا به سندرم تریچر کالینز، جهت تخفیف بر رنج این بیماران میباشد. امید است در آیندهای نزدیک، از تکنیکهای ژنتیک مولکولی بویژه ژن درمانی، بتوان این اختلال ژنتیکی را درمان کرد و اگر چنین تکنیکی کارساز باشد، میبایست در دوران جنینی این اختلال را در رحم مادر درمان کرد. به عبارتی جنین دارای سندرم تریچر کالینز، اما نوزاد متولد شده سالم خواهد بود.

References:
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. pp. 894, 1686.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews’ Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders.
- Conte, Chiara; Maria Rosaria D’Apice; Fabrizio Rinaldi; Stefano Gambardella; Federica Sanguiuolo; Giuseppe Novelli (27 September 2011).
- Posnick, Jeffrey C (1 October 1997). “Treacher Collins syndrome: Perspectives in evaluation and treatment”. Journal of Oral and Maxillofacial Surgery 55 (10): 1120–1133.
- Hertle, R W; Ziylan, S; Katowitz, J A (1 October 1993). “Ophthalmic features and visual prognosis in the Treacher-Collins syndrome.”. British Journal of Ophthalmology 77 (10): 642–645.
- Marszałek, B; Wójcicki, P; Kobus, K; Trzeciak, WH (2002). “Clinical features, treatment and genetic background of Treacher Collins syndrome.”. Journal of applied genetics 43 (2): 223–33.
- Dixon, Jill; Edwards, Sara J.; Gladwin, Amanda J.; Dixon, Michael J.; Loftus, Stacie K.; Bonner, Cynthia A.; Koprivnikar, Kathryn; Wasmuth, John J. (31 January 1996). “Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome”. Nature Genetics 12 (2): 130–136.
- Altug Teber, Özge; Gillessen-Kaesbach, Gabriele; Fischer, Sven; Böhringer, Stefan; Albrecht, Beate; Albert, Angelika; Arslan-Kirchner, Mine; Haan, Eric; Hagedorn-Greiwe, Monika; Hammans, Christof; Henn, Wolfram; Hinkel, Georg Klaus; König, Rainer; Kunstmann, Erdmute; Kunze, Jürgen; Neumann, Luitgard M; Prott, Eva-Christina; Rauch, Anita; Rott, Hans-Dieter; Seidel, Heide; Spranger, Stephanie; Sprengel, Martin; Zoll, Barbara; Lohmann, Dietmar R; Wieczorek, Dagmar (1 September 2004). “Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation”. European Journal of Human Genetics 12 (11): 879–890.
- Masotti, Cibele; Ornelas, Camila C; Splendore-Gordonos, Alessandra; Moura, Ricardo; Félix, Têmis M; Alonso, Nivaldo; Camargo, Anamaria A; Passos-Bueno, Maria (1 January 2009).
- Saadeh, Pierre B.; Chang, Christopher C.; Warren, Stephen M.; Reavey, Patrick; McCarthy, Joseph G.; Siebert, John W. (1 June 2008). “Microsurgical Correction of Facial Contour Deformities in Patients with Craniofacial Malformations: A 15-Year Experience”. Plastic and Reconstructive Surgery 121 (6): 368e–378e.
- Argenta, Louis C.; Iacobucci, John J. (30 June 1989). “Treacher Collins Syndrome: Present concepts of the disorder and their surgical correction”. World Journal of Surgery 13 (4): 401–409.
- Marres, HA; Cremers, CW; Marres, EH (1995). “Treacher-Collins syndrome. Management of major and minor anomalies of the ear.”. Revue de laryngologie – otologie – rhinologie 116 (2): 105–8.
- Nicholson, Nannette; Christensen, Lisa; Dornhoffer, John; Martin, Patti; Smith-Olinde, Laura (1 January 2011). “Verification of Speech Spectrum Audibility for Pediatric Baha Softband Users With Craniofacial Anomalies”. The Cleft Palate-Craniofacial Journal 48 (1): 56–65.
https://medlineplus.gov/genetics/condition/treacher-collins-syndrome/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام