سندرم بِسل هاگِن
Bessel-Hagen Syndrome

شاهین اسعدی (کارشناس ارشد ژنتیک)
کلیاتی از سندرم بِسل هاگِن
سندرم بِسل هاگِن که تحت عنوان اُستئوکندرومهای چندگانه ارثی نیز شناخته میشود، یک بیماری ژنتیکی است که در آن مبتلایان، تومورهای چندگانه خوشخیم (غیر سرطانی) استخوانی به نام اُستئوکندروم تولید میکنند. تعداد اُستئوکندرومها و استخوانهایی که در آنها مستقر هستند بهشدت در بین افراد مبتلا متفاوت است.
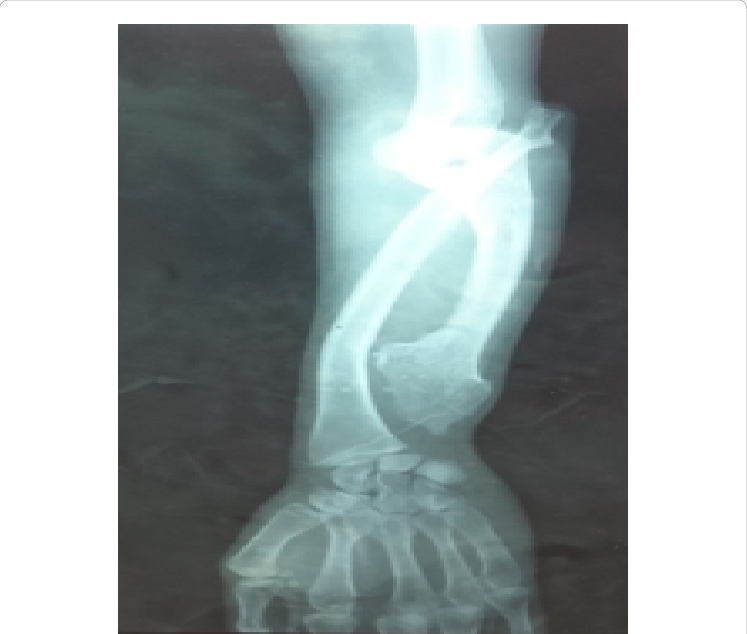
شکل 1: شماتیکی از اختلالات استخوانی در پای پسر مبتلا به سندرم بسل هاگن
علائم و نشانههای بالینی سندرم بِسل هاگِن
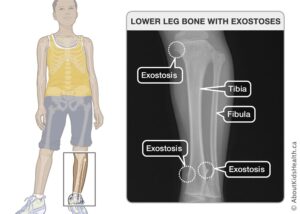
اُستئوکندرومها در هنگام تولد وجود ندارد، اما تقریباً 96 درصد از افراد مبتلا به این بیماری اُستئوکندرومهای چندگانه را در سن 12 سالگی توسعه میدهند. اُستئوکندرومها معمولاً در انتهای استخوانهای طویل و در استخوانهای مسطح مانند لگن و شانه قرار میگیرند.
شکل 2: تصاویری از اختلالات استخوانی (استئوکندروم) در مبتلایان سندرم بسل هاگن
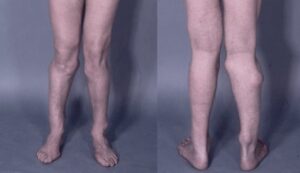
اُستئوکندرومهای متعدد میتوانند رشد استخوان را مختل کرده و باعث ایجاد اختلالات رشدی دستها، بازوها و پاها شوند که منجر به کوتاهی قد میشود. اغلب این مشکلات بر روی رشد استخوان اندام راست و چپ بهطور مساوی تأثیر نمیگذارند و در نتیجه اندامهای نامساوی (اختلاف طول اندام) ایجاد میشود. افتادن ساعد یا مُچ پا و توسعه غیرطبیعی مفصل ران (دیسپلازی هیپ) ناشی از اُستئوکندرومها است که میتواند به مشکلات راه رفتن و ناراحتی کلی منجر شود. اُستئوکندرومهای متعدد ممکن است در اثر درد، محدودیت حرکات مفصلی و فشار روی اعصاب، عروق خونی، نخاع و بافتهای اطراف اُستئوکندروم ایجاد شود.
اُستئوکندرومها معمولاً خوشخیم هستند، با این حال، در برخی موارد این تومورها بدخیم (سرطانی) میشوند. محققان تخمین میزنند که افرادی که دارای اُستئوکندرومهای چندگانه ارثی هستند در طول زندگی خود تا 20 برابر خطر ابتلا به اُستئوکندروم سرطانی (سارکوم) را دارند.
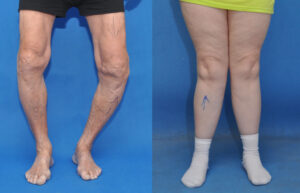
شکل 3: نمایی دیگر از اُستئوکندروم در پاهای مبتلایان سندرم بسل هاگن
علتشناسی سندرم بِسل هاگِن
سندرم بِسل هاگِن در اثر جهش ژن EXT1 که در بازوی بلند کروموزوم شماره 8 بهصورت 8q24.11 مستقر است و ژن EXT2 که در بازوی کوتاه کروموزوم شماره 11 بهصورت 11p11.2 مستقر است، ایجاد میشود. ژن EXT1 و ژن EXT2 دستورالعملهای لازم برای تولید پروتئینهای اگزوستوسین-1 و اگزوستوسین-2 را فراهم میکنند. دو پروتئین اگزوستوسین با یکدیگر همپوشانی دارند و یک کمپلکس ایجاد میکنند که در ساختار سلولی به نام دستگاه گُلژی یافت میشوند که آنزیمهای تازهتولیدشده و پروتئینهای دیگر را تغییر میدهد. در دستگاه گُلژی، اگزوستوسین-1 و اگزوستوسین-2، یک پروتئین به نام هپاران سولفات را اصلاح میکنند تا سلول بتواند از آن استفاده کند.
هنگامی که یک جهش در اگزوستیسین-1 یا اگزوستوسین-2 رخ میدهد، هپاران سولفات نمیتواند بهدرستی پردازش شود و غیرفعال خواهد بود. گرچه هپاران سولفات در بسیاری از فرآیندهای بدن دخیل است، اما هنوز مشخص نیست که عدم وجود این پروتئین در توسعه اُستئوکندرومها چه تأثیری دارد.
اگر این سندرم، ناشی از جهش در ژن EXT1 بوجود آید، این نوع اُستئوکندرومهای متعدد ارثی به نام نوع 1 نامیده میشوند. جهش در ژن EXT2 باعث ایجاد اُستئوکندرومهای متعدد ارثی نوع 2 میشود. در حالی که هر دو نوع 1 و 2 شامل چندین اُستئوکندروم هستند، با این حال، جهش در ژن EXT1 احتمالاً 55 تا 75 درصد موارد از همه موارد سندرم اُستئوکندروم چندگانه ارثی را ایجاد میکند و شدت علائم مرتبط با اُستئوکندروم در نوع 1 بهنظر میرسد بیشتر از نوع 2 باشد.
محققان تخمین میزنند که حدود 15 درصد از افراد مبتلا به سندرم بِسل هاگِن هیچ جهشی در ژن EXT1 یا ژن EXT2 ندارند. هنوز شناختهشده نیست که چرا اُستئوکندرومهای متعدد در این افراد ایجاد میشود.

شکل 4: نمای شماتیک از کروموزوم شماره 8 که ژن EXT1 در بازوی بلند این کروموزوم بهصورت 8q24.11 مستقر است
سندرم بِسل هاگِن از الگوی توارثی اتوزومال غالب پیروی میکند، بنابراین برای ایجاد این سندرم، یک نسخه از ژنهای جهشیافته EXT1 و EXT2 (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به این سندرم در حالت اتوزومی غالب برای هر بارداری احتمالی به میزان 50% است.

شکل 5: نمای شماتیک از کروموزوم شماره 11 که ژن EXT2 در بازوی کوتاه این کروموزوم بهصورت 11p11.2 مستقر است
فراوانی سندرم بِسل هاگِن
میزان بروز سندرم بِسل هاگِن حدود 1 در 000،5 نفر تخمین زده میشود. این بیماری بیشتر در برخی از جمعیتهای ایزوله رخ میدهد: میزان آن در جمعیت چاموروی گوام حدود 1 در 1000 نفر و در جمعیت هندوهای مانیتوبای کانادا حدود 1 در 77 نفر است.
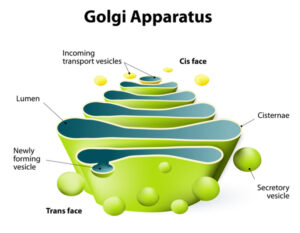
شکل 6: شماتیکی از ساختار دستگاه گُلژی
تشخیص سندرم بِسل هاگِن
سندرم بِسل هاگِن بر اساس یافتههای بالینی و کلینیکی مبتلایان و برخی آزمایشهای پاتولوژیکی و یا تصویربرداری رادیولوژیکی قابل تشخیص است. دقیقترین روش تشخیص این سندرم آزمایش ژنتیک مولکولی برای ژنهای EXT1 و EXT2 بهمنظور بررسی وجود جهشهای احتمالی است.
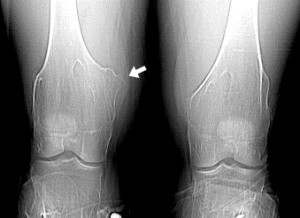
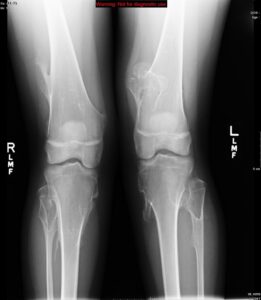
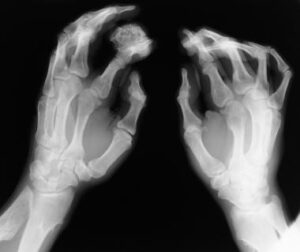
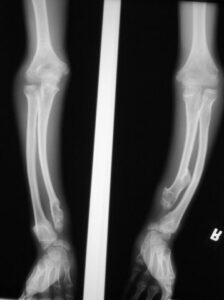
شکل 7: تصاویر رادیولوژیکی از اختلالات اسکلتی در سندرم بسل هاگن
مسیرهای درمانی سندرم بِسل هاگِن
استراتژی درمان و مدیریت سندرم بِسل هاگِن بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص ارتوپدی و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان متعارفی برای این سندرم وجود ندارد و تمامی اقدامات بالینی بهمنظور تخفیف رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند، ضرورت دارد.
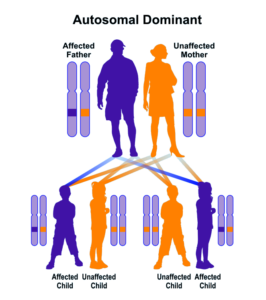
شکل 8: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم بسل هاگن از این الگو تبعیت میکند
منبع:
اسعدی شاهین، دکتر جلیلی امیر، دکتر نعمتی سهیل، مبلغ حسینی محمدحسین، جمالی مهسا، ولیزاده گلنساء، حبشیزاده مونا، حبیبی سعیده، عیوضوند عبداله، سادهدل سمانه، کتاب پاتولوژی در ژنتیک پزشکی جلد 15 (A-H)، صفحات 264-257، انتشارات کتب دانشگاهی عمیدی، تابستان 1398.
سندرم بال پروانهای Epidermolysis Bullosa (EB) Syndrome
ورود / ثبت نام