سندرم آلپورت
Alports Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، دکتر سید مجتبی محدث اردبیلی (متخصص ژنتیک پزشکی)، دکتر علی نظیرزاده (متخصص ژنتیک مولکولی)، الناز حیدری (دانشجوی کارشناسی ارشد ژنتیک)
نگارنده مسئول: شاهین اسعدی (Geneticist)


سندرم آلپورت یا نفریت ارثی، یک اختلال ژنتیکی است که با گلومرولونفریت، مرحله نهایی بیماری کلیوی و از دست دادن حس شنوایی همراه است. از اثرات سندرم آلپورت، میتوان به اختلالات چشم از جمله آبمروارید، قوز قرنیه، Lenticonus و همچنین تشکیل بلورهای شبکیه در ماکولا و حاشیه چشم اشاره کرد. هماچوری یا ادرار خونین و پروتئین در ادرار نیز از ویژگیهای سندرم آلپورت میباشند. این اختلال اولین بار توسط دکتر آرتور سیسیل آلپورت، پزشک فارغالتحصیل از دانشگاه ادینبورگ به سال 1927 در یک خانواده انگلیسی شناسایی شد.

علت شناسی سندرم آلپورت
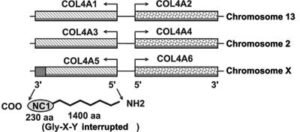
سندرم آلپورت، در اثر جهش در ژنهای COL4A3،COL4A4،COL4A5 و اکثر ژنهای دخیل در بیوسنتز پروتئین کلاژن ایجاد میشود. جهش در هر یک از این ژنها، باعث جلوگیری از تولید یا مونتاژ مناسب از شبکه کلاژن نوع چهارم که یک جزء ساختاری مهم از غشای پایه در کلیه، گوش داخلی و چشم است، میگردد.
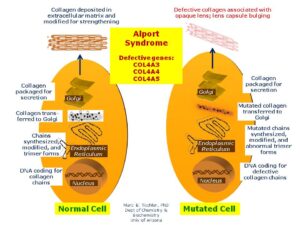
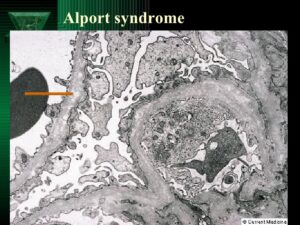
غشای پایه بهصورت سازههای ورق مانند نازک، بسیاری از سلولهای پشتیبان در بافتها را تشکیل میدهد. هنگامیکه جهش ژنتیکی در ژنهای مزبور رخ داد، از تشکیل الیاف کلاژن نوع چهارم جلوگیری میکند و همین امر موجب میشود تا غشای پایه از کلیهها، قادر به فیلتراسیون مواد زائد از خون و ادرار نباشد که در این صورت هماچوری یا خون در ادرار و پروتئین در ادرار شخص مبتلا به سندرم آلپورت مشاهده میشود. اختلالات کلاژن نوع چهارم در غشای پایه کلیه، منجر به زخم تدریجی کلیه میگردد و در نهایت نارسایی کلیهها را ایجاد میکند. افزایش ضخامت غشای پایه (به دلیل اسکار بافت) باعث ظاهر شدن بافت سبدی شکل به دلیل تفکیک از غشای پایه گلومرولی بهصورت لایهلایه از لامینای سلول میشود.
مطالعات محاسباتی تک مولکول در مولکول کلاژن نوع چهارم، تغییرات در ساختار و رفتار نانومکانیکی مولکولهای جهشیافته در این سندرم را نشان دادهاند. شایان ذکر است که این فرآیند، منجر به خمیدگی در مولکول پروتئین کلاژن نوع چهارم میشود.
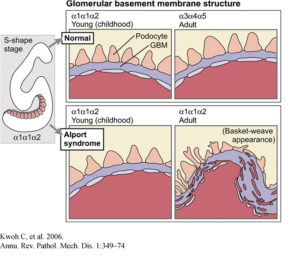
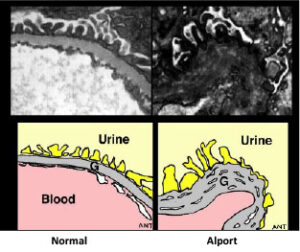
الگوهای توارثی و ژنتیک مولکولی سندرم آلپورت
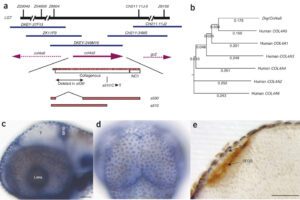
سندرم آلپورت میتواند الگوهای توارثی متفاوت بسته به نوع جهش ایجادکننده داشته باشد. اکثر افراد مبتلا به سندرم آلپورت (%85)، از الگوی توارثی وابسته به X با جهش در ژن COL4A5 تبعیت میکنند. ژن COL4A5 در بازوی بلند کروموزوم جنسی X بهصورت Xq22.2 مستقر است.
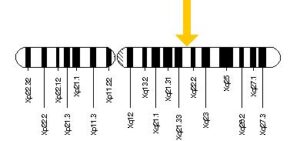
بسته به جنسیت افراد مبتلا به سندرم آلپورت، میزان شدت بیماری متفاوت خواهد بود؛ به عبارتی در مردان، چون دارای یک کروموزوم X هستند اگر ژن COL4A5 که در کروموزوم جنسی X واقع است، دچار جهش شود، بیشترین استعداد را برای مردان بهمنظور نارسایی کلیهها و اختلال گوارشی در پی خواهد داشت، اما زنان چون دارای دو کروموزوم جنسی X هستند، جهش در ژن مزبور کمترین استعداد را برای اختلالات کلیوی بوجود خواهد آورد و زنان مبتلا به سندرم آلپورت معمولاً خون در ادرار را نشان میدهند ولی نارسایی کلیوی را نخواهند داشت.
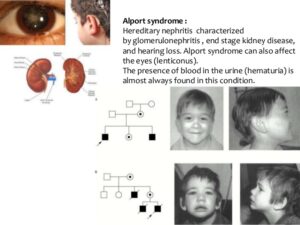
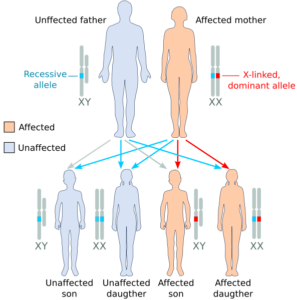
سندرم آلپورت همچنین میتواند با الگوی توارثی، اتوزومال مغلوب نیز به نسل بعد منتقل شود. اگر هر دو نسخه از ژن COL4A3 و COL4A4 که در بازوی بلند کروموزوم شماره 2 بهصورت 2q35.1 و 2q36.3 مستقر هستند، دچار جهش شوند، در نتیجه فرزند نسل بعد نیز به سندرم آلپورت مبتلا خواهد شد. در اغلب موارد ممکن است، پدر و مادر کودک مبتلا به سندرم آلپورت با الگوی توارثی اتوزومال مغلوب، بیماری را بروز ندهند و فقط بهعنوان حامل یک ژن تغییریافته از ژنهای ذکرشده باشند.
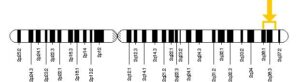
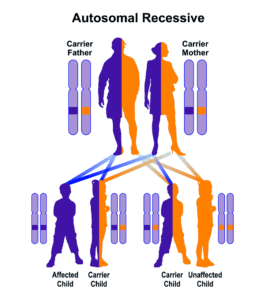
در گذشته، سندرم آلپورت را بهصورت اتوزومال غالب نیز معرفی کرده بودند. البته انتقال این بیماری بهصورت اتوزومال غالب نادر است و تنها در %5 از موارد بیماری میتواند وجود داشته باشد. نشانههای بالینی سندرم آلپورت با الگوی توارثی اتوزومال غالب، شبیه نشانههای بالینی این سندرم با الگوی توارثی وابسته به X است، با این حال تشدید نارسایی کلیوی در این بیماران، آهستهتر از الگوی توارثی وابسته به X میباشد.
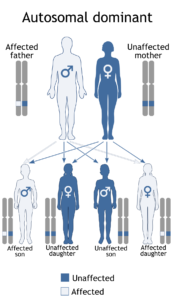
تشخیص سندرم آلپورت
برای تشخیص فرد مبتلا به سندرم آلپورت بایستی چندین معیار، رعایت گردد:
a) سابقه خانوادگی در نارسایی کلیوی و هماچوری یا خون در ادرار
b) هماچوری مداوم بدون عوامل نفروپاتی از قبیل بیماری غشاء پایه گلومرولی نازک، بیماری پلیکیستیک کلیه و یا نفروپاتی ایمونوگلوبین آلفا (IgA)
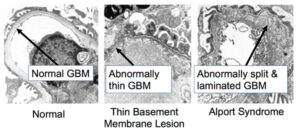
c) از دست دادن حس عصبی شنوایی دوطرفه در محدوده بسامدهای 2000-8000 HZ هرتز. از دست دادن حس عصبی شنوایی بهصورت تدریجی توسعه مییابد و معمولاً تا قبل از 30 سالگی آغاز میشود.
d) جهش در ژنهای COL4An که ضریب n بهصورت ,4,5 n=3 است.
e) ایمونوهیستوشیمی، شواهد فقدان کامل یا جزئی از اپیتوپ آلپورت در گلومرولی و یا غشای پایه اپیدرمی و یا هر دو را نشان دهد.
f) بروز اختلالات فراساختاری گسترده در غشای پایه گلومرولی در ضخامت خاص و یا نازک شدن یا تقسیم شدن این غشاء در بافتها
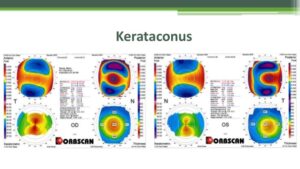
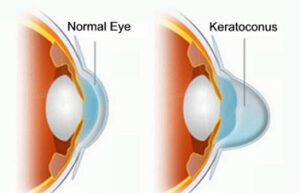
g) وجود ضایعات چشمی از جمله lenticonus قدامی، kerataconus، آبمروارید subcapsular خلفی، دیستروفی چندریختی خلفی و بلورهای شبکیه
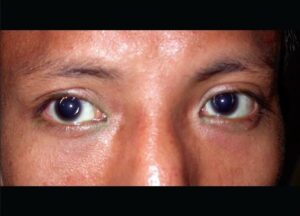

تصویر فوق دیدگان یک فرد مبتلا به سندرم آلپورت با اختلال چشمی kerataconus یا دوبینی حالت توهمی را نشان میدهد
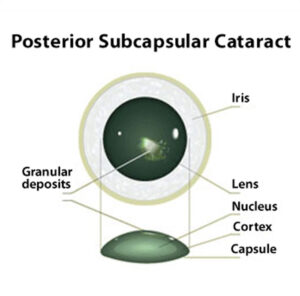
تصویر فوق، نمای شماتیک چشم با اختلال subcapsular خلفی را نشان میدهد
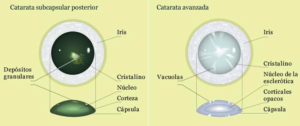
تصویر فوق، نمای شماتیک از چشم با اختلال آبمروارید زیر کپسول خلفی را نشان میدهد

تصویر فوق، دیدگان یک فرد مبتلا به اختلال آبمروارید زیر کپسول خلفی را در مقابل دیدگان چشم سالم نشان میدهد
h) پیشرفت تدریجی بیماری کلیوی و رسیدن به مرحله نهایی نارسایی کلیوی که حداقل در دو عضو خانواده مشهود گردد.
i) مشاهده ماکروترومبوسیتوپنیا یا گرانولوسیتیک اجزاء خونی در نمونه خون محیطی بیمار که مشابه به آنومالی May-Hegglin میباشد.
j) مشاهده لئومایوماتوزیس منتشر مری یا دستگاه تناسلی زنان یا هر دو
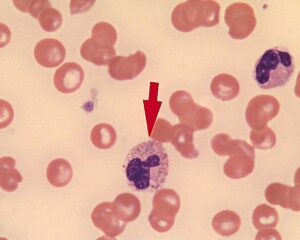
تصویر ماکروترومبوسیتوپنیا در گستره خون محیطی بیمار زیر میکروسکوپ

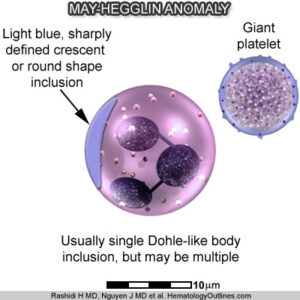
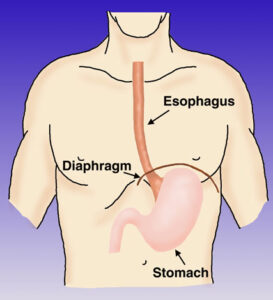
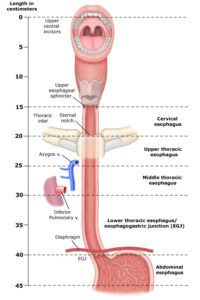
نمای شماتیک از اجزاء esophagus انسان
با این حال قطعیترین آزمایش جهت تشخیص سندرم آلپورت، آزمایش ژنتیک مولکولی برای ژنهای COL4A3،COL4A4،COL4A5 میباشد که بایستی بعد از آزمایش ژنتیک به این بیماران کارت ژنتیکی بالینی بهمنظور غربالگری سریع و آسان تحویل داده شود.
ایمونوهیستوشیمی سندرم آلپورت
ایمونوهیستوشیمی (IHC) شواهدی از سندرم آلپورت فرم وابسته به X را، ممکن است از بیوپسی پوست یا گلومرول کلیوی بدست آورد. در این پروسه، از آنتیبادی برای تشخیص وجود یا عدم وجود، Alpha3، Alpha5،Alpha4 در زنجیره کلاژن نوع چهارم استفاده میشود.
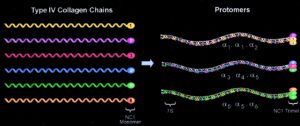
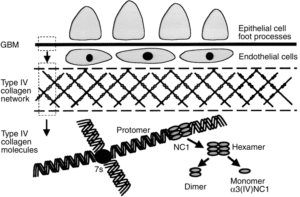
هر سه این زنجیره آلفا، در افراد سالم بهصورت طبیعی در غشای پایه گلومرولی فعالیت میکنند. در افراد مبتلا به سندرم آلپورت با الگوی توارثی وابسته به X، زنجیره Alpha5 بهطور ناکارآمد فعالیت کرده که منجر به مونتاژ کلاژن نوع چهارم بهصورت شکسته و یا خمیده میگردد. از میان این سه زنجیره آلفا، فقط زنجیره Alpha5 در سلولهای بافت پوست بیان میشود، بطوریکه مشخصه سندرم آلپورت وابسته به X در بیوپسی پوست صورت، عدم وجود زنجیره Alpha5 میباشد.
مسیرهای درمانی سندرم آلپورت
هیچ درمان شناختهشدهای برای این بیماری وجود ندارد؛ اما میتوان بیماران مبتلا به سندرم آلپورت را از نظر عوارض نارسایی کلیوی و دفع پروتئین از ادرار مدیریت کرد که اغلب برای جلوگیری از دفع پروتئین از طریق ادرار از مهارکنندههای ACE استفاده میشود.
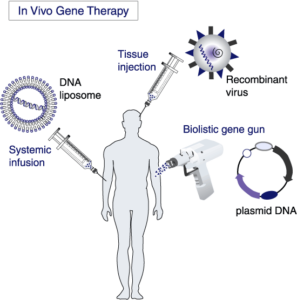
هنگامیکه نارسایی کلیه عارض شد، میتوان از دیالیز یا پیوند کلیه سالم برای جلوگیری از رنج بیماران استفاده کرد، اگرچه ممکن است خود این فرآیند نیز مشکلاتی را به وجود آورد و سیستم ایمنی بدن، عضو جدید را پس بزند. ژندرمانی بهعنوان یک مسیر درمانی سالم و ایدهآل، میتواند در سالهای آتی برای این بیماران مناسبتر باشد.
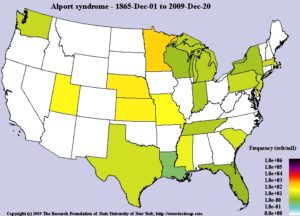
تصویر فوق، میزان فرکانس سندرم آلپورت در مناطق مختلف جهان از 1 دسامبر 1865 تا 20 دسامبر 2009 را نشان میدهد
References:
- Lagona E، Tsartsali L، Kostaridou S، Skiathitou A، Georgaki E، Sotsiou F (April 2008).
- Alport، AC (19 March 1927). “Hereditary familial congenital haemorrhagic nephritis”. BMJ 1 (3454): 504–506.
- Srinivasan M، Uzel SGM، Gautieri A، Keten S، Buehler MJ (2009). “Alport Syndrome mutations in type IV tropocollagen alter molecular structure and nanomechanical properties”. J. Structural Biology 168 (3): 503–510.
- The more frequent presentation of Alport Syndrome appear from mutations in the COL4A5 gene، which are inherited in an X-linked dominant form; see Jais JP et al.
- Pescucci C، Mari F، Longo I; et al. (May 2004). “Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene”. Kidney Int. 65 (5): 1598–603.
- Chen، Dilys; Jefferson، Barbara; Harvey، Scott J.; Zheng، Keqin; Gartley، Cathy J.; Jacobs، Robert M.; Thorner، Paul S. (October 2003).
- Cheungpasitporn W; Kaewpoowat Q; Suksaranjit P; Kittanamongkolchai W; Srivali N; Ungprasert P; Rangan Y (2012). “Autosomal Dominant Alport Syndrome Presenting as Proteinuria at Marine Corps Physical Fitness Test: A Case Report and Review.”. J Nephrol Therapeut S8.
- Gregory MC، Terreros DA، Barker DF، Fain PN، Denison JC، Atkin CL (1996). “Alport syndrome–clinical phenotypes، incidence، and pathology”. Contrib Nephrol 117: 1–28.
- Zhang KW، Colville D، Tan R; et al. (August 2008). “The use of ocular abnormalities to diagnose X-linked Alport syndrome in children”. Pediatr. Nephrol. 23 (8): 1245–50.
- Fausto، [ed. by] Vinay Kumar; Abul K. Abbas; Nelson (2005). Robbins and Cotran pathologic basis of disease. (7th ed.). Philadelphia: Elsevier/Saunders. p. 988.
https://medlineplus.gov/genetics/condition/alport-syndrome/
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام