
کارآیی بالینی دو ارزیابی سرولوژیک SARS-CoV-2

دکتر شاهرخ مستور تهرانی
پیشزمینه: ظهور کروناویروس سندرم حاد تنفسی ۲ (SARS-CoV-2) منجر به گسترش سریع ارزیابیهای سرولوژیک شده است، اگرچه اطلاعات اندکی در مورد کارایی بالینی آنها وجود دارد. در این مقاله ما سنجش تجاری CoV-2 IgG را با یکدیگر مقایسه کردهایم.
روشها: ۱۰۳ نمونه از ۴۸ بیمار که عفونت SARS-CoV-2 آنها از طریق PCR تأیید شده بود و ۱۵۳ نمونه کنترل با استفاده از ارزیابیهای سرولوژیک SARS-CoV-2 با استفاده از کیتهای Abbott و EURIIMMUN (EI) آنالیز شدند. با بررسی سوابق پزشکی، فاصله زمانی بروز علائم تعیین شد. حساسیت تشخیصی، اختصاصیت و همخوانی نتایج محاسبه گردید.
نتایج: در سنجش Abbott SARS-CoV-2، اختصاصیت تشخیصی بدین شرح بود:
99/4% (95% CI: 96/41 – 99/89%) و حساسیت 0/0% (95% CI: 0/00 – 26/47%) در کمتر از 3 روز پس از ظهور علائم، 30/0% (95% CI: 11/89 – 54/28%) در روزهای 7-3، 47/8%
(95% CI: 26/82 – 69/41%) در روزهای13-8 و 93/8% (95% CI: 82/80 – 98/69%) در 14 روز یا بیشتر. اختصاصیت تشخیصی برای سنجش EI به صورت زیر است: اگر نتایج مرزی مثبت در نظر گرفته شوند
94/81% (95% CI: 89/96-97/72%) و 96/7% (95% CI: 92/54 – 98/93%) اگر نتایج مرزی، منفی در نظر گرفته شود. حساسیت تشخیصی ۰/۰% (95% CI: 0/00 – 26/47%) را در < 3 روز،
25/0% (95% CI: 8/66-49/10%) در روزهای 7-3، 56/5% (95% CI: 34/49- 76/71%) در روزهای
3-7 و 85/4% (95% CI: 72/24- 93/93%) در ≥ 14 روز است البته اگر نتایج مرزی مثبت در نظر گرفته شود. همخوانی اندکی بین این دو سنجش 0/83 (95% CI: 0/75 – 0/91) بود.
نتیجهگیری: سنجش Abbott SARS-CoV-2 نسبت به سنجش EI نتایج مثبت کاذب و منفی کاذب کمتری داشت. اگرچه حساسیت تشخیصی در هر دو سنجش در طول ۱۴ روز ابتدایی بروز علائم، ضعیف بود.
مقدمه
رشد سریعی در تولید تستهای سرولوژیک برای (SARS-CoV-2)- پاتوژنی که عامل پاندمی کروناویروس ۲۰۱۹ (COVID-19) است- رخ داده است. در مقابل مجوز اجباریِ استفاده اضطراری (emergency use authorization; EUA) که بر آزمایشهای مولکولی برای SARS-CoV-2 حاکم است، FDA در ابتدا EUA را برای نظارت بر تستهای سرولوژی الزامی نمیدانست که همین امر به گسترش سریع آزمایشهای سرولوژیک SARS-CoV-2 با کارایی بالینی نامعلوم منجر شد. استفادههای پیشنهادی بسیاری از سرولوژی SARS- CoV-2 وجود دارد؛ از جمله مطالعات شیوع سرولوژیک (seroprevalence) و تعیین وضعیت ایمنی. بسیاری از مطالعات که سعی در بررسی این تستهای کاربردی داشتهاند، از کوچک بودن سایز نمونه به زحمت افتادند و یا در سرورهای پیش از چاپ بدون بازبینی منتشر شدهاند. برخی از انجمنهای حرفهای نیز توصیههایی را ارائه کردهاند، اما تاکنون در مورد اندیکاسیونهای اصلی سرولوژی اتفاق نظر وجود نداشته است؛ به عنوان مثال، انجمن بیماریهای عفونی آمریکا (IDSA) پیشنهاد میکند که سرولوژی میتواند در موارد زیر مفید باشد:
۱) بیمارانی با علائم بالینی که تا حد زیادی مطرحکننده وجود SARS-CoV-2 است اما تست RNA آنها منفی است.
2) انتخاب اهداکنندگان پلاسما پس از بهبودی از بیماری؛
3) ارزیابی پاسخهای واکسن؛
4) مطالعات اپیدمیولوژیک
هم IDSA و هم WHO به شدت نسبت به ارتباط سرولوژی مثبت با مصونیت (immunity) هشدار دادهاند.
از آنجایی که از پلاسمای پس از بهبودی با موفقیت در درمان بیماران COVID-19 استفاده شده است، فرض بر این است که آنتیبادیها در برابر SARS-CoV-2 محافظتکننده هستند. با این وجود، اختصاصیت اپیتوپ و تیتر موردنیاز برای دستیابی به مصونیت هنوز مشخص نشده است. IgG و/ یا IgM معمولاً در پاسخ آنتیبادی بیماری عفونی اندازهگیری میشوند، اگرچه IgA نیز در برخی از روشهای SARS-CoV-2 استفاده شده است. در زمان نگارش مقاله، از 12 روش سرولوژیکی که EUA دریافت کردهاند، 5 روش فقط IgG را اندازهگیری میکنند، 3 روش هم IgM و IgG را اندازهگیری میکنند و 4 روش هم کل ایمونوگلوبولینها را اندازهگیری میکنند. مطالعات آنتیبادی طولی (longitudinal) نتایج متناقضی را در مورد تیترهای IgM و IgG ضد SARS-CoV-2 را در طول دوره بیماری نشان داده است.
علاوه بر این، آنتیژن مناسب برای استفاده برای تشخیص آنتیبادیهای در گردش علیه SARS-CoV-2 هنوز تعریف نشده است. چندین تولیدکننده، سنجشهایی را طراحی کردهاند که آنتیبادیهای علیه پروتئین spike ویروس که واسطه ورود SARS-CoV-2 به سلولهای میزبان هستند یا پروتئین نوکلئوکپسید که یک پروتئین ساختاری بسیار ایمونوژنیک است را اندازهگیری میکنند. همولوژی قابلتوجهی بین SARS-CoV-2 و سایر کروناویروسهای فصلی وجود دارد و باعث میشود انتخاب آنتیژن برای ایمونواسیها برای برخورداری از اختصاصیت بالا بسیار مهم باشد.
تا به امروز هیچ مقایسهای از ELISAهای تجاری برای آنتیبادیهای SARS-CoV-2 در ادبیات تحقیق بررسیشده وجود نداشته است، به علاوه، به دلیل تقاضای زیاد برای این سنجشها در طی پاندمی، اعتبارسنجی برخی تولیدکنندگان مختصر شده است. در این مقـــــــــاله، ما عملکرد دو سنجش آنتیبادی IgG ضد SARS-CoV-2 از Abbott و EUROIMMUN (EI) را مقایسه کردهایم.
مواد و روشها
نمونههای آزمایش
این تحقیق توسط Institutional Review Board of Washington University in St. Louis ,MO تایید شد. از نمونههای باقیمانده بیمار که به درخواست پزشک برای آزمایش شمارش کامل خون در لولههای EDTA Vacutainer (BD) به آزمایشگاه بیمارستان Barnes Jewish ارسال شده بودند، استفاده شد. زیرمجموعهای از نمونههای سرم که در سال 2015 جمعآوری شده بود و قبل از ظهور SARS-CoV-2 در دمای 80- درجه سانتیگراد ذخیره شده بود، به عنوان نمونههای کنترل استفاده گردید.
با توجه به فقدان راهنمایی روشن از سوی انجمنهای تخصصی در مورد سودمندی روشهای سرولوژی، پروتکل اعتبارسنجی فرض میکند که هدف اصلی آزمایش برای غربالگری مبتنی بر جمعیت (یعنی مطالعات اپیدمیولوژیک یا غربالگری بیماران بدون علامت) است و برای بیمارانی که بیش از ۱۰ روز از شروع علائمشان گذشته و PCR آنها منفی است، انجام شده است. 103 نمونه از 48 بیمار مبتلا عفونت COVID-19 تأیید شده و 153 نمونه منفی فرض شده، مورد تجزیه و تحلیل قرار گرفت. نمونههای کنترل شامل موارد ذیل بودند:
80 بیمار علامتدار اما با PCR منفی برای SARS-CoV-2، 50 نمونه سرم جمعآوری شده و منجمد شده در سال 2015، قبل از ظهور SARS-CoV-2، 5 نمونه از بیماران مبتلا به کرونا ویروسهای دیگر که با آزمایش مولکولی تأیید شدند اما PCR آنها برای COVID-19 منفی بود (از جمله Coronaviruses HKU1 ، NL63 و 229E) و 4 نمونه از بیماران مبتلا به آنفلوانزا A یا B. ۱۴ نمونه دارای آنتیبادیهای بالقوه مداخلهکننده نیز استفاده شدند. 5 مورد از نظرCMV IgG ، 3 مورد از نظرEBV VCA IgG ، 3 مورد از نظرEBV VCA IgM ، 2 مورد از نظر EBV VCA IgG- IgM و 2 مورد از نظر فاکتور روماتوئید مثبت بودند.
اطلاعات بالینی
مدت زمان شروع علائم با بررسی پرونده الکترونیکی پزشکی توسط 2 ارزیاب مستقل و استنباط از یادداشتهای پزشک تعیین شد. علائم به صورت شکایت اصلی در مراجعه فعلی تعریف شده بود. علائم اولیه شامل علائم تنفسی (یعنی تنگی نفس)، سرفه، تب، از دست دادن چشایی یا بویایی، سردرد و گلودرد بود. شرایط پزشکی زمینهای که میتواند بر ایمنی هومورال تأثیر بگذارد در صورت امکان ثبت شد.
دستگاهها و آنالیز
نمونهها با استفاده از 2 ایمونواسی جداگانه مورد تجزیه و تحلیل قرار گرفتند. روش Abbott SARS-CoV-2 IgG بر اساس دستورالعمل سازنده بر روی Abbott Architect i2000 (Abbott Diagnostics) انجام شد. روش Abbott یک روش کیفی است که اتصال IgG به یک اپیتوپ ناشناخته از پروتئین نوکلئوکپسید
SARS-CoV-2 را تشخیص میدهد. روش EI SARS-CoV-2 IgG (EUROIMMUN) یک قالب پلیت با 96 چاهک است که طبق دستورالعمل سازنده بر روی یک
Inova QUANTALyser 240 (Inova Diagnostics Inc.) اتوماتیک انجام شد. روش EI، IgG ضد
SARS-CoV-2 با ویژگی علیه دومین S1 پروتئین spike ویروسی را تشخیص میدهد. هر دو روش با استفاده از یک کالیبراتور اختصاصی آزمایش، نسبت جذب نمونه به جذب کالیبراتور را گزارش میدهند. تفسیر نهایی مثبت بودن با نسبت بالاتر از مقدار آستانه تعیین میشود. سنجش Abbott به صورت مثبت (نسبت ≥ 1/4) یا منفی (نسبت < 1/4) و سنجش EI به صورت مثبت (نسبت ≥ ۱/۱)، حالت مرزی (نسبت < ۱/۱ تا ≥ 0/8) و منفی (نسبت < 0/8) تفسیر شد. کنترل کیفی (QC) که توسط سازنده ارائه میشود، در هر روز آزمایش مورد آزمایش قرار گرفت. برای مطالعات تداخل، بر اساس دستورالعملهای سازنده، شاخص همولیز، شاخص ایکتریک و لیپمیک با استفاده از یک آنالایزر Roche e 602 (Roche) تعیین گردید.
وجود یا عدم وجود عفونت SARS-CoV-2 با شناسایی RNA ویروسی SARS-CoV-2 از سوابهای نازوفارینژیال (NP)، سوابهای حفره اوروفارنژیال (OP) یا نمونههای مجرای تنفسی تحتانی در آزمایشگاه بیمارستان Barnes Jewish با سنجشهای اعتبارسنجی شده برای استفاده بالینی تأیید شد. به دلیل کمبود معرف، از چندین پلتفرم برای ارزیابی حضور SARSCoV-2 RNA اســــتفاده شـــــــــــــد. سنجش Quidel Lyra RT-PCR که پلی پروتئین غیرساختاری SARS-CoV-2 (pp1ab) را تشخیص میدهد روش اصلی استفاده شده (حد تشخیص copies/mL ۸۰۰) بود. برخی از بیماران بر اساس نتایج آزمایش مولکولی با استفاده از سنجش مولکولی Xpert Xpress SARS-CoV-2 (Cepheid) (حد تشخیص copies/mL ۲۵۰) و Simplexa COVID-19 Direct Assay با استفاده از LIAISON MDX (Diasorin) (تنها با استفاده از نمونههای مجرای تنفس تحتانی، حد تشخیص copies/mL 500) تأیید شدند.
نمونههای باقیمانده تا زمان آنالیز در دمای ˚ ۸۰- سانتیگراد و در مقادیر 500 میکرولیتری منجمد شد. هر نمونه طی 24 ساعت ذوب و آنالیز شد. از همان مقدار ۵۰۰ میکرولیتری برای هر دو سنجش استفاده شد.
اعتبارسنجی و دقت تست
مطالعات دقت با استفاده از نسخه اصلاحشده گایدلاین CLSI EP12-A2 انجام شد. برای مطالعات دقت، مواد QC (مثبت و منفی) که توسط Abbott و EI تهیه شده بود، مورد تجزیه و تحلیل قرار گرفت. با استفاده از پلاسمای کنترل منفی و سرم یک بیمار مثبت رقتهایی تا حد نزدیک به به cut-off مثبت هر دو سنجش، تهیه شد. تکرارپذیری با آنالیز 20 تکرار (replicate) در یک روز با روش Abott ارزیابی گردید. با توجه به محدود بودن دسترسی به معرف، تکرارپذیری با 10 تکرار در یک روز با روش EI ارزیابی شد. عدم دقت (imprecision) کل در طول 5 روز ارزیابی گردید. تداخل، برای همولیز، یرقان و لیپمی با استفاده از پلاسمای دو بیمار مثبت که با سرم یک بیمار منفی رقیق شده بود، ارزیابی شد.
آنالیز آماری
حساسیت و اختصاصیت تشخیصی برای هر سنجش با استفاده از آزمایش مولکولی به عنوان استاندارد طلایی محاسبه شد. برای سنجش EI، حساسیت و اختصاصیت با این فرض محاسبه شد که نتایج مرزی بر اساس گایدلاینهای ارائهشده به تولیدکنندگان توسط FDA، مثبت یا منفی تلقی گردد. با این حال، با توجه به نگرانی از وجود نتایج مثبت کاذب، نتایج مرزی در درجه اول به عنوان نتایج مثبت گزارش شدند. از زمان مثبت شدن PCR نیز برای محاسبه حساسیت و اختصاصیت استفاده شد. یک بیمار که دارای PCR منفی بود، اما از نظر بالینی COVID-19 + محسوب میشد و با هر دو ایمونواسی نیز مثبت بود؛ بنابراین، یک آنالیز اختصاصیت جداگانه برای این نمونه دارای عدم تطابق انجام شد. همخوانی نتایج با استفاده از Cohen’s Koppa و درصد توافق محاسبه شد. برای تعیین زمان بروز علائم، در تمام بیمارانی که پاسخ ایمنی بروز دادند، از یک رگرسیون لجستیک ۴-پارامتری سیگموئیدی، برای متناسب کردن دادهها و ربط دادن روزها(ی پس از ابتلا) به مثبت شدن نتیجه، استفاده گردید (بیمارانی که پاسخهای ایمنی نشان ندادند، از مطالعه حذف شدند). مدلسازی سیگموئیدی بر اساس کینتیک پاسخ ایمنی ایجادشده توسط 12 بیمار تحت نظارت متوالی، انتخاب شد. تمام آنالیزهای آماری با GraphPad Prism 8 انجام شد.
نتایج:
در سنجش Abbott SARS-CoV-2، اختصاصیت تشخیصی تام 99/4% (95% CI: 96/41 – 99/98%) بود و حساسیت تشخیصی در < 3 روز پس از ظهور علائم 0/0% (95% CI: 0/00 – 26/47%)، در ۷-۳ روز
30/0% (95% CI:11/89 – 54/28%) و در 13-8 روز 47/8% (95% CI: 26/82 – 69/41%) بود. در بیمارانی که ۱۴ روز یا بیشتر از شروع علائم آنها میگذشت حساسیت 93/8% (95% CI: 82/80 – 98/69%) بود. با استفاده از زمان بعد از مثبت شدن نتایج PCR حساسیت سنجش Abbott در ۳ روز پس از تست PCR، ۶/۴۷ (95% CI: 32/00 –63/58%)، 3-7 روز بعد 59/1% (95% CI: 36/35- 79/29%)، 8-13 روز بعد 69/6% (95% CI: 47/08- 86/79%) و بیش از 14 روز 81/3% (95% CI: 54/35- 95/95%) بود (شکل 1B). 12 نفر از 27 بیمار اگر به صورت همزمان، با PCR تست میشدند، با استفاده از تست سرولوژیک نیز مثبت بودند. در بیمارانی که به طور همزمان با تست سرولوژی و تست مولکولی ارزیابی شدند (در روز ۰)، میانگین زمان بروز علائم در بیمارانی که تست سرولوژیک مثبت داشتند 12/2 روز (میانه= ۱۴ روز) بود در حالی که میانگین زمان بروز علائم در بیمارانی با تست سرولوژیک منفی، ۳/۳ روز (میانه= ۲ روز) بود.
اختصاصیت تشخیصی برای سنجش EI به این صورت بود: اگر نتایج مرزی مثبت در نظر گرفته شوند 94/8% (95% CI: 89/96- 97/72%) و 96/7% (95% CI: 92/54 – 98/93%) اگر نتایج مرزی منفی در نظر گرفته شود. (شکل 2A). حساسیت تشخیصی در < 3 روز ۰/۰% (95% CI: 0/00 – 26/47%)، در روزهای 7-3، ۰25/0% (95% CI: 8/66 – 49/10%)، در روزهای 13-8، 56/5% (95% CI: 34/49- 76/81%) و در روز ۱۴ 85/4% (95% CI: 72/24- 93/93%) بود، البته اگر نتایج مرزی مثبت در نظر گرفته شود. برمبنای زمان بعد از مثبت شدن نتایج PCR حساسیت سنجش EI در < 3 روز، 38/1% (95% CI: 23/57– 54/36%)، 7-3 روز بعد 63/6% (95% CI: 40/66- 82/80%)، 8-13 روز بعد 69/6% (95% CI: 47/08- 86/89%) و بیش از 14 روز 75/0% (95% CI: 47/62- 92/73%) بود (شکل 2B). محاسبات حساسیت با در نظر گرفتن نتایج مرزی به عنوان نتایج منفی محاسبه شد که در جدول تکمیلی ۱ قابل مشاهده است.

4 نتیجه ناسازگار بین روشهای سرولوژی و آزمایش مولکولی وجود داشت. از 3 بیماری که پاسخ آنتیبادی را تا روز چهاردهم در هر دو سنجش سرولوژیک نشان ندادند، 2 نفر تحت شیمیدرمانی برای لوسمی بودند. نفر سوم نقص ایمنی مشخصشدهای نداشت اما قبلاً مبتلا به نقص 3-هیدروکسی آسیل کوآنزیم A دهیدروژناز زنجیره بلند تشخیص داده شده بود. یک بیمار (دایره خاکستری با X، شکل 1 و 2) به وسیله تست مولکولی، منفی تشخیص داده شد بود، اما با هر دو ایمونواسی مثبت بود. این بیمار با بیش از 10 روز علائم کلاسیک COVID (تب، سردرد، از دست دادن چشایی و بویایی) و چند بار مواجهه با اعضای خانواده که مبتلا به COVID تأیید شده، بودند؛ از نظر بالینی به عنوان COVID-19+ شناخته میشد. اگر این بیمار از آنالیز کنار گذاشته شود، در صورت مثبت در نظر گرفتن نتایج مرزی، اختصاصیت تشخیصی سنجش Abbott و EI به ترتیب ۱۰۰% (95% CI: 97/6- 100/0%) و 95/39% (95% CI: 90/74 – 98/13%) بود.

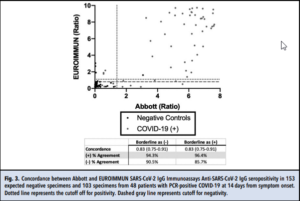
توافق کلی درصد مثبت بین روشهای EI SARS-CoV-2 و Abbott، در صورتی که نتایج مرزی به ترتیب منفی یا مثبت در نظر گرفته شوند، 94/3% و 96/4% است. (شکل ۳). در صورت منفی در نظر گرفتن نتایج مرزی، توافق کلی درصد منفی 90/5% و در صورت مثبت در نظر گرفتن 85/7% بود.
همخوانی کلی نتایج بدون در نظر گرفتن نحوه قضاوت نتایج مرزی، 0/83 (95% CI: 0/75- 0/91) بود. دو نمونه از بیمارانی که از نظر SARS-CoV-2 منفی بودند، در ارزیابی EI به صورت ضعیف مثبت بودند. هر دو بیمار دچار علائم حاد تنفسی و عفونتهای مربوط به سایر پاتوژنها بودند. سومین عدم تطابق مثبت در EI مربوط به نمونه جمعآوری شده قبل از پاندمی بود.
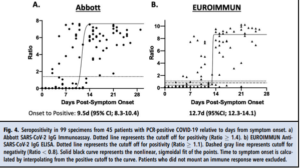
کینتیک سروکانورشن IgG با استفاده از نمونههای 12 بیمار با نتایج سریالی ارزیابی شد (شکل 1 تکمیلی). تناسب رگرسیون غیرخطی دادهها از شروع علائم در مقابل آشکار شدن سیگنال، نتیجه IgG مثبت را با 9/5 روز (95% CI: 8/3- 10/4) در سنجش Abbott SARS-CoV-2 و 12/7روز (95% CI: 12/3- 14/1) در سنجش EI نشان داد (شکل 4). رقتهای سریالی یک نمونه در هر دو سنجش، یک منحنی خطی را نشان داد (شکل 2 تکمیلی). سنجش Abbott تا تیتر ۱:۱۶ مثبت باقی ماند، در حالی که EI تا 1:32 مثبت باقی مانده و در 1:64 به صورت مرزی بود.
برای سنجش Abott SARS-CoV-2، تکرارپذیری و عدم دقت کل سیگنال برای QC مثبت و پولد سرم بیمار < 2% و توافق نتایج کیفی 100% بود (جدول تکمیلی 2). تکرارپذیری و عدم دقت کل EI بر اساس CVبه ترتیب 3/73% و 4/16%، برای کنترل مثبت و 9/61% و 12/25% برای پولد سرم بیمار و توافق نتایج کیفی 100% بود. برای هر دو سنجش، حداقل تداخل ناشی از همولیز، یرقان و لیپمیا وجود داشت (جدول تکمیلی 3). در سنجش Abbott هیچگونه انتقالی (carryover) از نمونهای با سیگنال بالا به نمونههای منفی مشاهده نشد (جدول تکمیلی 4).
بحث
در این مقاله، دو سنجش سرولوژیک تجاری برای تشخیص آنتیبادیهای SARS-CoV-2 در پلاسمای انسان ارزیابی شدند. ما دریافتیم که سنجش Abbott نسبت به سنجش EI حساسیت و اختصاصیت تشخیصی بیشتری دارد، اگرچه همپوشانی فاصلههای اطمینان نشان میدهد که این یافته از لحاظ آماری معنادار نیست و مطالعات بیشتری برای تأیید این موضوع لازم است. با وجود این، با بررسی و تأیید نتایج به وسیله تشخیص مولکولی، مشخص شد که سنجش EI نسبت به سنجش Abbott با نتایج منفی کاذب و مثبت کاذب بیشتری همراه است. نکته قابل توجه آنکه هیچکدام از این دو سنجش از حساسیت کافی برای تشخیص قابلاعتماد آنتیبادیها قبل از روز چهاردهم پس از بروز علائم، برخوردار نبودند. بنا بر اطلاعات ما، این مطالعه اولین مقایسه از این نوع استن که منطبق بر گایدلاین های اعتبار بخشی بوده و پلتفرمهای تجاری را بررسی میکند.
با توجه به الزامات تنظیمی سهلگیرانه FDA در مورد تستهای سرولوژیک SARS-CoV-2، ضروری است که آزمایشگاههای بالینی از سختگیری در اعتبارسنجی این ارزیابیها حمایت کنند و تعیین کنند که آیا کارایی یک سنجش آنگونه که در بستهبندی آن درج شده دقیق است یا خیر؛ برای مثال تنها ۹ نمونه از ۸ بیمار برای تعیین حساسیت تشخیصی گزارش شده در بستهبندی تست EI SARS-CoV-2 استفاده شده بود. در مقابل CLSI توصیه میکند حداقل از نمونههای ۵۰ بیمار تأییدشده باید استفاده شود. جالب توجه است که ما به ۳ بیمار برخوردیم که در روز چهاردهم پس از عفونت، در هر دو سنجش، پاسخ ایمنی سرولوژیک نشان ندادند. این مسئله با موارد درجشده در بستهبندی تست Abbott که ادعای حساسیت تشخیصی ۱۰۰ درصدی دارد، مغایر است. نتایج به دست آمده در مطالعه ما احتمالاً با نتایج تولیدکنندگان متفاوت است، زیرا اکثر جمعیت بیمار ما با چندین سناریوی بالینی همپوشان در بیمارستان بستری شده بودند. این مسئله اهمیت آزمایش بیمارانی را که به طور کامل از عفونت بهبود یافتهاند و بیماران بستری با چندین همابتلایی از جمله نقص ایمنی را نشان میدهد؛ به علاوه، برخی از تولیدکنندگان و آزمایشگاهها از زمان مثبت بودن SARS-CoV-2 PCR برای تعریف حساسیت سرولوژی در زمانهای مختلف بیماری بهجای زمان شروع علائم استفاده کردهاند. در این مطالعه، ما این دو روش را برای محاسبه حساسیت مقایسه کردیم. ما دریافتیم که حساسیت در نقاط زمانی ابتداییتر، هنگامی که با مثبت بودن PCR محک زده شود نسبت به زمانی که با بروز علائم محک زده شود، بالاتر است. با این حال، این احتمالاً تخمین بیش از حد حساسیت است، زیرا برخی از بیماران دیر مراجعه کرده بودند و در هنگام اولین آزمایش PCR در مراحل پیشرفته بیماری خود بودند. به عنوان شاهدی بر این مسئله، زمان متوسط شروع علائم در بیماران با PCR و نتایج مثبت سرولوژیک در همان روز آزمایش 14 روز بود. با این وجود، قضاوت از 14 روز پس از شروع علائم و 14 روز از نتایج مثبت PCR، حساسیت مشابهی را ارائه میدهد. مهم است که آزمایشگاهها هنگام محاسبه حساسیت و ارائه این اطلاعات به ارائهدهندگان مراقبتهای بهداشتی، چگونگی زمان تعیین نتایج مثبت را تعریف کنند. همچنین نتایج ما حساسیت پایین تشخیص آنتیبادیهای SARS-CoV-2 قبل از ۱۴ روز پس از بروز علائم را در هر دو سنجش نشان میدهد. این مسئله دلیلی است بر اینکه که وضعیت سرولوژیک نباید تا 14 روز بعد از شروع علائم ارزیابی شود و تأیید میکند که روشهای مولکولی باید روش اصلی برای تشخیص COVID-19 باشد.
اختصاصیت کم خصوصاً در مورد سنجش EI نگرانکننده است، زیرا از سنجش سرولوژیک برای غربالگری جمعیت و مطالعات اپیدمیولوژیک استفاده میشود. جالب توجه است که هیچیک از نتایج مثبت کاذب در سنجش EI مربوط به بیماران مبتلا به کرونا ویروس فصلی نبوده است و سه مورد مثبت کاذب مربوط به نمونههای جمعآوری شده در سال 2015 بوده است. این مسئله ممکن است حداقل تا حدی به دلیل تعداد بالای بیماران دارای آنتیبادی علیه کروناویروسهای فصلی که دارای همولوژی قابلتوجهی با SARS-CoV-2 هستند، باشد. تعداد موارد تأییدشده فعلی COVID-19 در ایالات متحده بیش از 1/03 میلیون نفر است که منجر به شیوع تخمینی تقریباً 0/32% شده است. در نتیجه، یک آزمایش سرولوژیک با اختصاصیت بالا برای دستیابی به ارزش پیشبینیکننده مثبت بالا (PPV) ضروری است. از آنجا که شیوع افراد با آنتیبادی مثبت SARS-CoV-2 در ایالات متحده در حال حاضر نامعلوم است، FDA عملکرد پیشبینیشده سنجشهای تولیدکنندگان را بر اساس شیوع ۵% خلاصه کرده است. با فرض شیوع ۵% و بر اساس نتایج گزارششده در این مقاله، PPV سنجش Abbott و EI به ترتیب 88/7% و45/9% است که اهمیت وجود اختصاصیت بالا را برجسته میسازد. با این حال، FDA همچنین اذعان میکند که در جمعیتهای با شیوع کم (مانند افراد بدون علامت و مطالعات غربالگری جمعیت)، احتمالاً یک آزمایش آنتیبادی بهتنهایی برای تشخیص مثبت واقعی از مثبت کاذب کافی نیست؛ به عنوان شاهدی بر این امر، کاهش شیوع به 0/5%، PPV سنجش Abbott SARS-CoV-2 را به 44% و EI را به 7/5% کاهش میدهد.
IDSA توصیه میکند که بیماران با علائم بالینی مطابق با COVID-19 اما با تست مولکولی منفی برای SARS-CoV-2 ممکن است توسط سرولوژی تشخیص داده شوند. در این مطالعه، فقط 1 نفر از 80 بیمار که دارای علائم و PCR منفی بود، توسط هر دو ایمونواسی مثبت بود. در حالی که فراوانی چنین بیمارانی با PCR منفی و سرولوژی مثبت ممکن است با بزرگتر شدن اندازه نمونه، افزایش یابد؛ مشاهدات ما همچنین نشان میدهد که سناریوی بالینی ارائهشده توسط IDSA ممکن است رویدادی با احتمال کم باشد. این مسئله این استدلال را که آزمایش مولکولی RNA ویروسی باید استاندارد طلایی برای تشخیص عفونتهای COVID-19 باشد را بیش از پیش تقویت میکند. برای ارزیابی کامل کاربرد تشخیص سرولوژیکی که توسط IDSA پیشنهاد شده است، مطالعات بزرگتری لازم است.
ما دریافتیم که زمان مثبت شدن سنجش سرولوژی Abbott که یک اپیتوپ نوکلئوکپسید ویروسی را تشخیص میدهد، در مقایسه با سنجش EI که اپی توپ ضد پروتئین spike ویروسی تشخیص میدهد؛ کوتاهتر است. این مسئله با مطالعات پیشین در مورد پاسخ سلول B به SARS-CoV مطابقت دارد، مبنی بر اینکه پاسخ ایمنی همورال معمولاً ابتدا در برابر نوکلئوکپسید ویروسی و به دنبال آن پاسخ در برابر پروتئین spike ویروسی ایجاد میشود؛ بنابراین، اینکه فکر کنید نوکلئوکپسید ویروسی ممکن است هدف بهتری برای تشخیص زودهنگام ایمونوگلوبولینها باشد، وسوسهانگیز است. با این حال، هم IDSA و هم CDC استفاده از سرولوژی را اکیداً برای تشخیص عفونت حاد SARS-CoV-2 توصیه نمیکنند که این مسئله ارتباط بالینی این یافته را محدود میکند. محدودیت دیگر سنجشهای سرولوژیک از نظر بالینی، اهمیت ناشناخته آنتیژنهای هدف قرارگرفته برای تشخیص است. تابهحال هنوز مشخص نیست که آیا آنتیبادی علیه نوکلئوکپسید SARS-CoV-2 یا پروتئین spike نوترالیزان هستند و محافظت ایجاد میکنند یا خیر. علاوه بر این، از آنجا که هر دو سنجش Abbott و EUROIMMUN سنجش کیفی هستند و هیچکدام برای تعیین کمی آنتیبادیهای ضد SARSCoV-2 تأیید نشدهاند، هنوز کاربرد آنها برای اهداکنندگان پلاسمای پس از بهبودی تعریف نشده است.
یک محدودیت مطالعه ما این است که شروع علائم به طور ذهنی توسط پزشکان گزارش شده و این اطلاعات با مرور دستی پرونده پزشکی بازیابی شده است؛ به عنوان مثال، بیماران ارجاع داده شده از خانه سالمندان به دلیل اختلالات شناختی، در یادآوری تاریخها ضعف داشتند. این مسئله ممکن است بر تخمین شروع بیماری در مطالعه ما و کینتیک پیشبینیشده تغییرات IgG در طی دوره بیماری تأثیر بگذارد. لذا، این نتایج کینتیکی، نیاز به اندازهگیری سریالی در برخی بیماران COVID+ را برجسته میسازد و نشان میدهد که یک نتیجه منفی واحد، لزوماً عفونت را رد نمیکند.
در نهایت اینکه، در این مطالعه سنجش Abbott SARS-CoV-2 حساسیت و اختصاصیت بالاتری از سنجش EI SARS-CoV-2 را نشان داد. حساسیت هر دو روش در طی 14 روز ابتدایی پس از شروع علائم ضعیف است و این نشان میدهد که برای تشخیص، نامناسب هستند. همانطور که روشهای جدید برای سنجش SARS-CoV-2 ظهور میکنند، برای ارزیابی عملکرد این سنجشها باید مطالعات اعتبارسنجی قوی انجام شود.
برگردان از:
Clinical Performance of Two SARS-CoV-2 Serologic Assays
Clinical Chemistry 66:8 (2020)
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام