microRNA و سرطان
(بخش دوم)
زهرا اصغری لالمی (دانشجوی دکتری ژنتیک مولکولی دانشگاه آزاد پرند)
سرطان نتیجهی خروج سلولها از مسیرهای درست تنظیمی، تکثیری و تمایزی است. خودکارآمدی[1] در سیگنالهای رشد، غیرحساس شدن به سیگنالهای مهارکنندهی رشد، اجتناب از مرگ سلولی برنامهریزیشده، پتانسیل نامحدود تکثیر، حفظ رگزایی و تهاجم بافتی و متاستاز، منجر به بدخیم شدن تومور میشوند. تحقیقات گسترده نشان داده است که microRNAها نقش اساسی در شروع، پیشرفت و متاستاز سرطان بازی میکنند (1 و 2). سطوح بیان microRNAها در تومورها میتواند تنظیم مثبت یا منفی در مقایسه با بافت نرمال داشته باشد و microRNAهای مختلف بهطور مستقیم در ایجاد تومور بهوسیلهی فعالیت بهعنوان انکومیر[2] یا سرکوبگر تومور[3] دخیل باشند (3). انکومیرها در بدخیمیهای بافتهای مختلف حضور دارند و اغلب در مناطقی از ژنوم که دچار حذفشدگی، مضاعفشدگی و یا موتاسیون شدهاند، یافت میشوند. در میان آنها میتوان به خوشهی miR-17-92 استناد کرد که اولین جایگاه microRNA انکوژنیک توصیف شده بود (4). در مقابل miR-a34 یک microRNA مهم با فعالیت سرکوب تومور میباشد که میتواند بهطور مستقیم بهوسیلهی P53 ترانس فعال شود. این عمل دوگانه را میتوان به زمینههای سلولی خاص که یک microRNA را در معرض تنظیم رونویسی متمایز قرار میدهد و/یا به اهداف مختلف RNA منسوب کرد (5). برخی microRNAها فنوتیپ انکوژنی را از طریق کاهش بیان ژنهای سرکوبکنندهی توموری و یا ژنهای تنظیمکنندهی تمایز سلولی و مرگ برنامهریزیشده ایجاد میکنند و برخی دیگر با هدف قرار دادن mRNAهای پروتوانکوژنیک و خاموش کردن آنها منجر به کاهش روند سرطانی شدن میشوند. برهمکنش microRNAها با ژنهای هدف، نقش آنها را در رشد، مرگ برنامهریزیشده، تمایز و تکثیر سلولی مشخص کرده و عملکرد مستقیم microRNAها را در سرطان تأیید میکند.
میتوان از بیان microRNAهای غالب برای طبقهبندی سرطانها در گروههایی با ویژگیهای متفاوت مثل نوع سلول و سببشناسی استفاده کرد. استفاده از microRNA برای طبقهبندی تومور بهمراتب مناسبتر از mRNA میباشد، این امر به علت جفتشدگی ناقص بین microRNA و mRNAهای هدف است، از این رو ریزسنجهای microRNA میتوانند بیان چند صد ژن و در نتیجه مسیرهای متعدد را در یک نمونه شناسایی کنند، در حالیکه تنها به مقادیر کمی از RNA کل نیاز دارند. ساختار microRNA و نحوهی عملکرد آنها نشان میدهد که بسیاری از microRNAها در نمونههای سرطانی بهصورت غیرطبیعی بیان میشوند. علاوه بر این، تفاوتهای عملکردی بین انواع تومورها و مراحل مختلف سرطان با بیان microRNAها مرتبط است. تفاوت در بیان microRNAها در سرطانهای مختلف میتواند به علت تفاوتهای موجود بین منشأ سلول سرطانی و بافت استرومایی اطراف آن باشد. تغییر در بیان microRNA از طریق کاهش بیان ژنهای ضروری درگیر در تکثیر یا بقای سلول، منجر به تشکیل تومور میشود. البته این به آن معنا نیست که microRNAها بهطور مستقیم در پیشروی سرطان یا تومورزایی نقش داشته باشند. هرچند هنوز بهطور کامل مشخص نشده است که بیان تغییریافتهی microRNAها پیامد حالت پاتولوژیکی سرطان است یا اینکه سرطان عامل مستقیم این تغییرات بیانی است، با این وجود تغییرات زیادی در سلولهای سرطانی رخ میدهد که میتوانند در یک مسیر مستقیم یا غیرمستقیم، بیان microRNA را تحت تأثیر قرار دهند. بازآرایی ژنومی، ناهنجاری ژنهای microRNA یا پروتئینهای درگیر در ساخت آنها، اختلال در تنظیم اپیژنتیک microRNA و موتاسیونهای ژنی نمونههایی از این تغییرات هستند، با این حال قرار گرفتن microRNAها در نواحی ژنومی مرتبط با سرطان یا نواحی شکننده، خود یک عامل مهم تغییر بیان microRNAها در سلولهای توموری است. علاوه بر این تحت تأثیر موتاسیون، ویژگیهای اتصالی microRNAها و mRNAها تغییر یافته و این برهمکنشهای تغییریافته microRNA:mRNA منجر به ناقص شدن فرآیندهای ترجمهای میشود. قابل ذکر است که عوامل اپیژنتیکی میتوانند microRNA را از طریق متیله کردن بیش از حد[4] یا تغییرات هیستونی[5] غیرفعال کنند و از طرف دیگر microRNA میتواند بهعنوان یک تنظیمکنندهی ژنتیکی عوامل فوق مطرح گردد. بیان افزایشیافتهی microRNA در سلولهای سرطانی ممکن است حاصل ازدیاد یا عدم کنترل یک فاکتور رونویسی یا دمتیله شدن[6] جزایر CpG در نواحی پروموتر ژنی باشد. حذفهای ژنی، خاموشکنندههای خارج ژنی یا عدم بیان فاکتورهای رونویسی در سلولهای سرطانی باعث کاهش بیان microRNAهای مهارکنندهی توموری میشود. محصولات microRNA پس از رونویسی و از طریق توالیهای اطراف microRNA پیشساز کنترل میشوند؛ بنابراین احتمالاً موتاسیونهای ایجادشده در توالیهای اطراف microRNA پیشساز بر روی پردازش microRNA تأثیر مستقیم گذاشته، منجر به کاهش بیان microRNA میشوند.
اولین مطالعهای که microRNAها را به سرطان مرتبط میکند در سال 2002 روی بیماران لوکمی لنفوسیتی مزمن انجام شد. کالین[7] و همکارانش مشاهده کردند که در حدود 65 درصد از این بیماران، خوشــــــههای ژنهای
15a-miR و 16-1-miR تنظیم منفی یا حذف شده است. درواقع نقش سرکوبگری توموری آنها پیشنهاد شد. متعاقباً همان گروه نقشهی درصد قابلتوجهی از miRها را تهیه کردند و دیدند که 52/5 درصد از آنها در نواحی پیوسته با سرطان و یا شکننده[8] واقع شدهاند (6). در سال 2004 آنالیز microchip برای مشخص کردن ژنهای microRNA در انسان و موش استفاده شد. Microchip استفاده شده حاوی 245، microRNA مختلف بود و این نتایج با تکنیکهای RT-PCR و Northern blot هم تأیید شدند. این ابزارها میتوانند برای آنالیز بیان microRNA در بافتهای سالم و بیمار و همچنین تغییر در الگوی بیان microRNA مورد استفاده قرار گیرند (7). در سال 2005 اولین گزارش که عدم تنظیم مناسب 29 عدد microRNA را در سرطان پستان بیان میکرد، منتشر گردید (8). الگوهای بیان microRNA بهشدت تنظیمشده میباشند و این مولکولها نقش عمدهای را در تکثیر، آپوپتوزیس و تمایز دارند. مطالعات متعدد نشان داده است که نمای بیان microRNA در بافتهای طبیعی متفاوت از بافتهای توموری بوده و نیز در بین انواع تومورها متفاوت میباشد. بهعنوان مثال نوع خاصی از microRNA که 203-miR نام دارد بهوسیلهی مکانیسمهای ژنتیکی در بسیاری از اشکال توموری سرطان خون بهخصوص لوکمی میلوژن مزمن و بعضی از انواع لوکمیهای لنفوسیتی حاد، خاموش میباشد. خاموشی یا عدم رونویسی از 203-miR سبب فقدان تنظیم نوعی ژن جهشیافته سرطانی با عنوان ABL1 و پروتئین رمزگذاری شدهی آن با عنوان BCR-ABL1 میشود (9).
بنابراین تغییرات مشاهده شده در کارنامهی microRNA در سرطان میتواند درنتیجهی: 1- انواع مکانیسمهای مخرب در ژنها (حذفها، تقویتها یا جهشهای ژنهای microRNA)، 2- تنظیم رونویسی (خاموشی اپیژنتیک، عدم تنظیم فاکتورهای رونویسی)، 3- تنظیم پس از رونویسی (عدم تنظیم مسیر بیوژنز microRNA) رخ دهد (10 و 11).
عملکردهای microRNA در سرطان
با توجه به اینکه هدف microRNA در سلول میتواند یک بازدارندهی تومور باشد، در سلولهای سرطانی دیده شده که این دسته microRNAها دچار افزایش بیان شده، در نتیجه بازدارندههای تومور در این سلولها کاهش پیدا میکنند که میتواند یکی از دلایل توموری شدن این سلولها باشد، این microRNAها را oncomiR مینامند (12)؛ بهعنوان مثال 21-miR در اکثر سرطانها دچار افزایش بیان میشود که بازدارندههای توموری از جمله PDCD4، PTEN و TPM1 در بین اهداف 21-miR هستند (13). در مقابل microRNAهایی هستند که هدفشان پروتوانکوژنهایی هستند که در سلولهای سرطانی دچار کاهش یا حذف میشوند. به این ترتیب پروتوانکوژنها در سلولها بالا میروند، بهعنوان مثال 7-Let در اکثر سلولهای سرطانی دچار کاهش بیان میشوند که پروتوانکوژن RAS یکی از اهداف مهم این microRNA است (14). مطالعات نشان دادهاند که در برخی سرطانها، آن دسته از microRNAها که هدفشان DNA متیل ترانسفراز است دچار کاهش بیان میشود که منجر به کاهش بازدارندهی تومور میشوند، همینطور microRNAهایی که هدفشان خاموشکنندهی کروماتین است هم دچار کاهش بیان شده و باعث پایین آمدن بازدارندهی تومور میشوند. دانشمندان این دسته microRNAها را که در سلولهای سرطانی دچار کاهش بیان میشوند، tsmiR مینامند (15). بیان بالای یک microRNA– مثلاً به دلیل مضاعف شدن ژن- بیان ژن هدف آن را کاهش میدهد که این ژن هدف در سلول سرطانی ژن بازدارندهی تومور است و بیان پایین یک microRNA– مثلاً به دلیل حذف شدن ژن- بیان ژن هدف آن را افزایش میدهد که این ژن هدف در سلول سرطانی یک انکوژن است (16).
microRNAها ویژگیهای بارز سلولهای سرطانی را تحت تأثیر قرار میدهند
تغییرات افزایشی یا کاهشی بیان برخی microRNAها که منجر به روند سرطانی میشوند، از طریق ایجاد تداخل با تنظیمکنندههای چرخهی سلولی، رشد سلول را تحت تأثیر قرار میدهند. microRNAها یکی از انواع تنظیمکنندههای اصلی مرگ برنامهریزیشده در فرآیند تومورزایی هستند و بقای سلولهای سرطانی با دستکاری این microRNAها کنترل میشود، از طرف دیگر سلولهای سرطانی ویژگی نامیرایی خود را با حفظ تلومرها از طریق تنظیم مثبت تلومراز و زیاد شدن طول تلومرها بدست میآورند. ناهنجاری microRNA یکی از عوامل فعالیت زیاد تلومراز در تومورها است. یک سری از microRNAها در گذار اپیتلیال- مزانشیمی که یکی از مراحل اساسی در هجوم تومور و متاستاز میباشد، درگیرند. قابل ذکر است که بیش از نیمی از microRNAها در مناطق ناپایدار ژنومی یا جایگاههای شکنندهی کروموزومها که با سرطانهای متنوعی همراه هستند بهصورت خوشهای قرار گرفتهاند. جابهجایی کروموزومی یک نشانگر حیاتی ناپایداری ژنومی است. microRNAها نقش مهمی را در تنظیم پاسخهای ایمنی در سرطان ایفا میکنند. microRNAهای ویروسی متنوعی در برهمکنش ویروس- میزبان در تومورزایی درگیرند، برای مثال SV40 که منجر به سرطان در جوندگان میشود دو microRNA کد میکند که رونوشت آنتیژن T ویروس را مورد هدف قرار داده و منجر به برش آن میشود. از آنجاییکه یک نقش microRNAها تجزیه و مهار ترجمهای mRNA است، آنزیمهای پردازشکنندهی microRNAیی Dicer و Drosha نهتنها در بلوغ microRNA نقش دارند، بلکه از طریق کاهش بیان خود، مانع از ایجاد رگ بهوسیلهی ژنهای از کنترل خارجشدهی مربوط به رگزایی میشوند (17).
مکانیسمهای اختلال تنظیمی microRNA در سرطان
در طول دههی گذشته روشن شده است که بیان microRNA در بدخیمیهای انسان دارای اختلال تنظیمی است. مکانیسمهای زیربنایی شامل اختلالات کروموزومی، تغییرات کنترلکنندهی رونویسی، تغییرات اپیژنتیک و نقص در سیستم تکامل و بیوژنز microRNA هستند.
1-1: قویت یا حذف ژنهای microRNA
بیان غیرطبیعی microRNA در سلولهای سرطانی در مقایسه با سلولهای طبیعی، اغلب منجر به تغییرات ژنومیک در تعداد کپیهای microRNA و مکانهای ژنی میشود (تقویت، حذف و یا انتقال). کشف اولیهی تغییر محل ژن microRNA، از دست دادن خوشهی ژنی 1-16/ miR15a در کروموزوم 14q13 است که اغلب در سلول B بیماران لوکمی لنفوسیتی مزمن مشاهده میشود (18). در سرطان ریه اغلب منطقهی حفاظتشدهی 33q5، 143-miR و 145-miR حذف شده، منجر به کاهش بیان هر دو microRNA میشود (19). در مقابل تقویت خوشهی ژنی 92-17-miR در سلول B لنفوما (20) و سرطانهای ریه (21) مشاهده شده است و انتقال این خوشهی ژنی همچنین در لوکمی لنفوبلاستی حاد سلولهای T مشاهده شده است که منجر به بیان بیش از حد این microRNAها در این سرطانها میشود (22). فرکانس بالای تغییرات ژنومی در جایگاه microRNA توسط وضوح بالای مبتنی بر آرایهی هیبریداسیون ژنومی مقایسهای در 227 نمونه سرطان تخمدان انسان، سرطان پستان و ملانوما تأیید شده است (23). بیشتر تحقیقات گستردهی ژنومی نشان دادند که بسیاری از ژنهای microRNA در نواحی ژنومی مرتبط با سرطان واقع شدهاند. این مناطق میتواند یک منطقهی حذفشده هتروزیگوتی باشد که میتواند ژن سرکوبگر تومور را حفظ کند یا یک منطقهی تقویتی باشد که ممکن است حاوی انکوژنها یا محلهای شکننده یا نواحی انفصال مشترک باشد (24). بهطور کلی، این یافتهها پیشنهاد میکند که بیان غیرطبیعی microRNA در سلولهای سرطانی میتواند از تقویت یا حذف ناشی از نواحی ژنومی خاص شامل ژنهای microRNA باشد.
2-1: کنترل رونویسی microRNAها
بیان microRNA شدیداً توسط فاکتورهای مختلف رونویسی کنترل شده است، بنابراین بیان غیرطبیعی microRNA در سرطان میتواند به علت اختلال تنظیمی بعضی از فاکتورهای کلیدی رونویســــــی مثل C-MYC و P53 باشد. ادانل و همکاران[9] (25) کشف کردند که C-myc غالباً در بسیاری از سرطانها تنظیم مثبت برای تنظیم تکثیر سلولی و آپوپتوزیس دارد. فعالیت و رونویســــــــی از انکوژن خوشهی 92-17-miR از طریق اتصال به عناصر E-Box در پروموتر 92-17-miR انجام میشود. مطابق با نقش انکوژنیک آنها، C-myc همچنین فعالیت رونویسی از microRNAهای سرکوبگر تومور مثل30-miR15a، miR- 29 ، miR و خانوادهی Let-7 را سرکوب میکند (26). گوشال و همکاران[10]، تنظیـــــم متقابل C-myc و سرکوبگر تومور 122-miR را در سرطــــــان هپاتوسلولار (کبدی) دریافتند. C-myc، بیان 122-miR را از طریق ارتباط با پروموتر آنها سرکوب میکند و 122-miR بهطور غیرمستقیم رونویسـی C-myc را از طریق هدف قرار دادن E2F1 و Tfdp2 مهار میکند؛ بنابراین، اختلال در این حلقهی بازخوردی بین 122-miR و C-myc امری ضروری برای پیشرفت سرطان هپاتوسلولار است (27). دیگر کاربرد حلقهی بازخوردی microRNA- C-myc، اختلال تنظیمی در سرطان هپاتوســــــــــلولار است. C-myc بهطور مستقیم به پروموترهای ژنهای p5-a148-miR / p3-363-miR متصل میشود و بیان آنها را سرکوب میکند. القای تومور سلولهای کبدی بهوسیلهی ترویج پیشرفت از فاز G1 به S است. بهنوبهی خود، p5-a148-miR، بیان C-myc را بهطور مستقیم هدف قرار میدهد و مهار میکند، درحالیکه p3-363-miR، توسط هدف قرار دادن مستقیم پروتئاز 28 مخصوص یوبیکویتیـــــنه کردن، C-myc را بیثبات میکند (28). 34-miR-p53 مثال دیگری از محور تنظیمی است که نشان میدهد فاکتور تنظیمی رونویسی بیان microRNA، برای میانجیگری سرکوب تومور، چطور عمل میکند (29). P53 یک سرکوبگر تومور کدکننده بهوسیلهی ژن Tp53، یکی از رایجترین ژنهای جهشیافته در سرطانهای انسان است. بیان تنظیمی P53 از بسیاری از ژنها، از جمله ژنهای microRNA، تشکیل یک شبکهی P53 پیچیده برای تنظیم پیشرفت چرخهی سلولی و آپوپتوزیس میدهد. مشابه با فنوتیپهای میانجیگر-P53، خانوادهی 34-miR از جمله c/b/a34-miR که به توقف چرخه سلولی، پیری و آپوپتوزیس سلولی منجر میشوند، در سرطان رایج هستند (30) که دلالت بر P53 و 34-miR در همان مسیر تنظیمی دارند. این فرضیه توسط اورن لب[11] (31) و مندل لب[12] (32) تأیید شد که نشان دادند که P53 میتواند بیان a34-miR بهمنظور آپوپتوزیس از طریق اتصال مستقیم به پروموتر ژن a34-miR را القا کند. بهنوبهی خود، a34-miR بیان P53 را از طریق هدف قرار دادن SIRT1، یک تنظیم منفی P53 از طریق دآسیله کردن گسترش میدهد (33). مطالعات بیشتر نشان داد که P53 عملکرد آنها را از طریق تنظیم بیان طیف وسیعی از microRNAها مثل 605-miR (34)، 1246-miR (35) و 107-miR (36) انجام میدهد. علاوه بر C-myc و P53 که دو مورد از بیشترین میزان مطالعات فاکتورهای رونویسی هستند، فاکتورهای رونویسی بیشتری بهمنظور تنظیم بیان microRNA یافت شـــــدهاند. برای مثال 223-miR ترجیحاً در سیستم خونساز با عملکردهای حیاتی در پیشرفت ردهی میلوئیدی بیان میشود و بیان آنها را در تومورهای متعدد از جمله سرطان هپاتوسلولار و لوکمی میلوئیدی حاد سرکوب میکند (AML) (37). فوکایو و همکاران[13] (38) دریافتند که بیان ژن 223-miR توسط فاکتورهای رونویسی میلوئیدی PU.1 و C/EBPs سرکوب میشود. فازی و همکاران[14] (39) کشف کردند که 223-miR و فاکتورهای رونویسی NFI-A و C/EBPα تشکیل یک مدار کوچک برای کنترل تمایز گرانولوستیک انسان را میدهند. این دو فاکتور رونویسی برای اتصال به پروموتر 223-miR رقابت میکنند. NFI-A، 223-miR را در سطوح پایین حفاظت میکند، در حالیکه رتینوییک اسید ناشی از C/EBPα، NFI-A را برای بیان تنظیمی 223-miR جایگزین میکند؛ بنابراین، بیان microRNA توسط فاکتورهای متعدد برای حفظ رونویسی طبیعی تنظیم شده است و اختلال در نظم آنها منجر به ایجاد تومور میشود.
3-1: اختلال تنظیمی تغییرات اپیژنتیکی
تغییرات اپیژنتیک یکی از ویژگیهای شناختهشده در سرطان است، از جمله هیپومتیلاسیون DNA ژنومی گسترده، هیپرمتیلاسیون نابجای DNA از ژنهای سرکوبگر تومور و اختلال در الگوهای اصلاح هیستون. اعتقاد بر این است که microRNAها، مشابه ژنهای کدکنندهی پروتئین، همچنین مستعد ابتلا به مدولاسیون اپیژنتیک هستند (40)؛ بهعنوان مثال فازی و همکاران (41) کشف کردند که بیان 223-miR بهصورت اپیژنتیکی توسط AML1/ETO، یک پروتئین ترکیبی همراه با AML رایج است که از طریق متیلاسیون CpG خاموش میشود. بررسیهایی توسط سایتو و همکاران[15] (42) انجام شد که نشان دادند، 17 عدد از 313 microRNAهای انسان بیش از سه برابر در سلولهای سرطان مثانه T24 پس از درمان بهطور همزمان با متیلاسیون DNA و مهارکنندههای استیلاسیون هیستون تنظیم مثبت دارند. در میان این microRNAها، 127-miR، در CpG جاسازی شده است و فاقد بیان در سلولهای سرطانی است. بهطور قابلتوجهی بیان پس از درمان بالا بود که با تنظیم منفی پروتوانکوژن BCL6 همراه بود. این نتایج نشان میدهد که دمتیلاسیون DNA و هیستون داستیلهی مهاری میتواند بیان microRNAها را فعال کند که ممکن است بهعنوان سرکوبگرهای تومور فعال شوند. با استفاده از روش مشابه، لوجامبیو و همکاران[16] (43) کشف کردند که خوشهی a148-miR و b/c34-miR موضوعی برای خاموشی مرتبط با هایپرمتیلاسیون اختصاصی در سلولهای سرطانی هستند. علاوه بر این، احیای این microRNAها در سلولهای سرطانی تحرک آنها را مهار میکند، رشد تومور را کاهش میدهد و تشکیل متاستاز در بدن را مهار مینماید. بهطور مشابه، کاهش بیان 1-9-miR و p5-145-miR به ترتیب به هیپرمتیلاسیون DNA در سرطانهای پستان، ریه و رودهی بزرگ منسوب میشوند (44). شواهد بالا نقش تنظیم اپیژنتیک در بیان microRNA در طول پیدایش تومور را نشان میدهد و یادآوری میکند که متیلاسیون نابجای DNA و استیلاسیون هیستون ژنهای microRNA میتواند بهعنوان نشانگرهای زیستی مفید برای تشخیص و پیشآگهی سرطان به کار گرفته شود.
4-1: نقص در مکانیسم بیوژنز microRNA
همانطور که در بخشهای قبل توضیح داده شد، بیوژنز microRNA توسط چندین آنزیم و پروتئین تنظیمی مانند Drosha، Dicer، DGCR8، پروتئینهای آرگونات و Exportin-5 ماهرانه کنترل شده است که اجازه میدهد بلوغ microRNA از پیشسازهای microRNA اولیه درست انجام شود؛ بنابراین، جهش یا بیان نابجای هر جزء از ماشین بیوژنز microRNA میتواند منجر به بیان غیرطبیعی microRNA شود. Drosha و Dicer دو تا از آنزیمهای RNAse III اندونوکلئازی کلیدی در بلوغ microRNA هستند که مسئول تشکیل Pre-miRNA و miRNA دوپلکس میباشند. تحقیقات اخیر نشان داد که هر دوی این آنزیمها در تومورهای خاص دچار اختلال در تنظیم هستند. تامسون و همکاران[17] (45) دریافتند که یک بخش بزرگ از microRNA در مرحلهی پردازش- دورشا تنظیم میشود و این تنظیم تأثیر عمدهای روی بیان microRNA در طول توسعهی جنینی و در سرطان دارد. والز (46) و همکاران[18] گزارش کردند که DGCR8 و دورشا جهشهای تعویضی/ حذفی تک نوکلئوتیدی در 15 درصد از 534 تومور ویلمز را دارند که منجر به کاهش قابلتوجه بیان از خانوادهی بالغ Let-7a و 200-miR میشود. با توجه به اختلال در نظم دایسر، مشاهده شد که دایسر 1 در سلولهای سرطان رودهی بزرگ اختلال ایجاد میکند که باعث کسب ظرفیت بیشتری برای آغاز و متاستاز تومور میشود (47). علاوه بر این سطوح بالای mRNA دورشا و دایسر در سرطان تخمدان با افزایش متوسط بقا همراه است (48) و بهطور معکوس، کاهش قابلتوجه بیان دایسر با کاهش بقای بیمار در ارتباط است (49). همبستگی مثبت بین کاهش سطوح mRNA دایسر و کاهش بیان Let-7 با بقای بعد از عمل نامطلوب همچنین توسط کاروب و همکاران[19] (50) در بیماران سرطان ریه کشف شد. پروتئینهای آرگونات اجزای کاتالیزوری ضروری RISC هستند و نقش مرکزی در فرآیندهای خاموش کردن RNA دارند. بهطور مشابه با دایسر و دورشا، اختلال ایجادشده در نظم پروتئینهای آرگونات در سرطان رخ میدهد، برای مثال، ژن EIF2C1/hAgo1 انسان اغلب در تومورهای ویلمز کلیه از دست میرود (51). بیان پروتئینهای آرگونات انسان (Ago) بهصورت وابسته به سلول تنظیم میشود؛ بهعنوان مثال، سطوح بیان Ago2 در سرطان معدهی اولیه و متاستاز غدد لنفاوی متناظر نسبت به این موضوع در افراد سالم قابلتوجهتر هستند (52)، در حالیکه بیان Ago2 پایینتر، متناظر با کاهش در بهرهوری از RNAi در ملانوما در مقایسه با ملانوسیتهای اولیه است (53). Exportin-5 (XPO5) یک پروتئین متصل به RNA دو رشتهای است که صادرات هستهای از Per-miRNA به درون سیتوپلاسم را وساطت میکند. ملو و همکاران[20] دریافتند که ژن XPO5 جهشها را در یک زیرمجموعه از تومورهای انسانی با میکروساتلایت بیثبات غیرفعال میکند. در CRC سلولهای HCT-15 و DLD-1، درج یک “A” در اگزون 32 سبب ایجاد کدون خاتمه زودرس میشود، در نتیجه منجر به جهش تغییر چارچوب و تولید نسخهی ناقص از پروتئین میشود. این XPO5 کوتاهشده، عملکرد خود را برای صادرات Pre-miRNA از دست میدهد؛ بنابراین Pre-miRNAها در هسته به دام افتاده، منجر به کاهش پردازش microRNA میشوند. مهمتر از همه، بازسازی معکوس عملکردهای XPO5، اختلال در صادرات Pre-miRNAها ایجاد میکند و ویژگیهای سرکوبگر تومور دارد (54). در کارسینومای هپاتوسلولار، همچنین مشاهده شد که شکستن XPO5 باعث انتقال Pre-miRNA از هسته به سیتوپلاسم میشود که بهوسیلهی ERK کیناز به XPO5 فسفریله میشود.
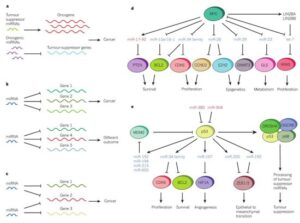
شکل 1: نقش microRNA در مسیرهای ایجاد سرطان (55)
منابع:
- N. Pencheva and S. F. Tavazoie, “Control of metastatic progression by microRNA regulatory networks,” Nature Cell Biology, vol. 15, no. 6, pp. 546–554, 2013.
- Y. Li, A. Ahmad, D. Kong, B. Bao, and F. H. Sarkar, “Targeting microRNAs for personalized cancer therapy,” Medical Principles and Practice, vol. 22, pp. 415–417, 2013.
- D. Wang, J. Gu, T. Wang, and Z. Ding, “OncomiRDB: a database for the experimentally verified oncogenic and tumorsuppressive microRNAs,” Bioinformatics, 2014.
- L. He, J. M. Thomson, M. T. Hemann et al., “A microRNA polycistron as a potential human oncogene,” Nature, vol. 435, no. 7043, pp. 828–833, 2005.
- JW. Nam, OS. Rissland, D. Koppstein, and et al, “Global analyses of the effect of different cellular contexts on microRNA targeting,” Molecular Cell, vol. 53, pp. 1031–1043, 2014.
- G. A. Calin, C. D. Dumitru, M. Shimizu et al., “Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia,” Proceedings of the National Academy of Sciences of the United States of America, vol. 99, no. 24, pp. 15524–15529, 2002.
- Davison TS, Johnson CD, Andruss BF. Analyzing micro-RNA expression using microarrays. Method Enzymol 2006; 411:14-34.
- Kerscher AE, Slack FJ. Oncomirs-microRNAs with a role in cancer. Nature 2006; 6:259-69.
- Babashah S, Sadeghizadeh M, Rezaii Tavirani M, Farivar S, Soleimani M. Aberrant microRNA expression and its implications in the pathogenesis of leukemia. Cellular Oncology. 2012; 35(5) 317-334.
- P. Lopez-Serra and M. Esteller, “DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer,” Oncogene, vol. 31, no. 13, pp. 1609–1622, 2012.
- Z. Wang, H. Yao, S. Lin et al., “Transcriptional and epigenetic regulation of humanmicroRNAs,” Cancer Letters, vol. 331, no. 1, pp. 1–10, 2013.
- Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Gen Develop 2005; 15 : 563-8.
- Si ML, Zhu S, Wu H. miR-21-mediated tumor growth. Oncogene 2007; 26:2799– 803.
- S. R. Viswanathan, G. Q. Daley, and R. I. Gregory, “Selective blockade of microRNA processing by Lin28,” Science, vol. 320, no. 5872, pp. 97–100, 2008.
- Croce CM. Causes and consequences of microRNA dysregulation in cancer, Nat Rev Genet 2009;10:704-14.
- Si ML, Zhu S, Wu H. miR-21-mediated tumour growth. Oncogene 2007;26: 2799-803.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74.
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2002; 99: 15524–15529.
- Calin GA, Croce CM. MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene 2006; 25: 6202–6210.
- Tagawa H, Seto M. A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia 2005; 19: 2013–2016.
- Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65: 9628–9632.
- Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol 2010; 12: 372–379.
- Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A et al. MicroRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA 2006; 103: 9136–9141.
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 2004; 101: 2999–3004.
- O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005; 435: 839–843.
- Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM et al. Widespread microRNA
in hepatocellular cancer: role of E2F1 and transcription factor dimerization partner 2. Hepatology 2014; 59: 555–566.
- Han H, repression by Myc contributes to tumorigenesis. Nat Genet 2008; 40: 43–50.
- Wang B, Hsu SH, Wang X, Kutay H, Bid HK, Yu J et al. Reciprocal regulation of microRNA-122 and c-Myc Sun D, Li W, Shen H, Zhu Y, Li C et al. c-Myc-MicroRNA functional feedback loop affects hepatocarcinogenesis. Hepatology 2013; 57: 2378–2389.
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130–1134.
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ 2010; 17: 193–199.
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N et al. Transcriptional activation of miR-34a contributes to p53 mediated apoptosis. Mol Cell 2007; 26: 731–743.
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26: 745–752.
- Yamakuchi M, Lowenstein CJ. MiR-34, SIRT1, and p53: The feedback loop. Cell Cycle 2009; 8: 712–715.
- Xiao J, Lin H, Luo X, Luo X, Wang Z. miR-605 joins p53 network to form a p53: miR-605:Mdm2 positive feedback loop in response to stress. EMBO J 2011; 30: 524–532.
- Zhang Y, Liao JM, Zeng SX, Lu H. p53 downregulates Down syndrome-associated DYRK1A through miR-1246. EMBO Rep 2011; 12: 811–817.
- Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT et al. p53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci USA 2010; 107: 6334–6339.
- Eyholzer M, Schmid S, Schardt JA, Haefliger S, Mueller BU, Pabst T. Complexity of miR-223 regulation by CEBPA in human AML. Leuk Res 2010; 34: 672–676.
- Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y et al. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell 2007; 129: 617–631.
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005; 123: 819–831.
- Han L, Witmer PD, Casey E, Valle D, Sukumar S. DNA methylation regulates MicroRNA expression. Cancer Biol Ther 2007; 6: 1284–1288.
- Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 2007; 12: 457–466.
- Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA et al. Specific activation of microRNA-127 with downregulation of the proto oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9: 435–443.
- Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 2008; 105: 13556–13561.
- Donzelli S, Mori F, Bellissimo T, Sacconi A, Casini B, Frixa T et al. Epigenetic silencing of miR-145-5p contributes to brain metastasis. Oncotarget 2015; 6: 35183–35201.
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 2006; 20: 2202–2207.
- Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 2015; 27: 286–297.
- Iliou MS, da Silva-Diz V, Carmona FJ, Ramalho-Carvalho J, Heyn H, Villanueva A et al. Impaired DICER1 function promotes stemness and metastasis in colon cancer. Oncogene 2014; 33: 4003–4015.
- Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med 2008; 359: 2641–2650.
- Faggad A, Budczies J, Tchernitsa O, Darb-Esfahani S, Sehouli J, Müller BM et al. Prognostic significance of Dicer expression in ovarian cancer-link to global microRNA changes and oestrogen receptor expression. J Pathol 2010; 220: 382–391.
- Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci 2005; 96: 111–115.
- Dome JS, Coppes MJ. Recent advances in Wilms tumor genetics. Curr Opin Pediatr 2002; 14: 5–11.
- Zhang J, Fan XS, Wang CX, Liu B, Li Q, Zhou XJ. Up-regulation of Ago2 expression in gastric carcinoma. Med Oncol 2013; 30: 628.
- Völler D, Reinders J, Meister G, Bosserhoff AK. Strong reduction of AGO2 expression in melanoma and cellular consequences. Br J Cancer 2013; 109: 3116–3124.
- Melo SA, Moutinho C, Ropero S, Calin GA, Rossi S, Spizzo R et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 2010; 18: 303 315.
- Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482(7385):347-55.
[1] Self-sufficiency
[2] Oncomir
[3] Tumor-Supresor
[4] Hyper methylation
[5] Histon modification
[6] Demethylation
[7] Caline
[8] Fragile
[9] O Donnell
[10] Ghoshal
[11] Oren Lab
[12] Mendell lab
[13] Fukao
[14] Fazi
[15] Saito
[16] Lujambio
[17] Tamson
[18] Walz
[19] Karube
[20] Melo
نقش mi-RNAها در پاتوژنز و درمان بیماری لوپوس اریتماتوس سیستمیک
Micro M.RNA، مارکری برای تشخیص سرطان
پلیمورفیسمهای تکنوکلئوتیدی در درمان و تحقیقات سرطان
برای دانلود فایل pdf بر روی لینک زیر کلیک کنید
ورود / ثبت نام