تشخیص آزمایشگاهی اختلالات کمی و کیفی کمبود فاکتور یک انعقادی
اکبر درگلاله1، حسن مروتی2
1: گروه هماتولوژی و طب انتقال خون، دانشگاه علوم پزشکی ایران
2: عضو هیئت علمی مرکز تحقیقات واکسن و سرمسازی رازی
اختلالات کیفی فیبرینوژن
در اختلالات کیفی فیبرینوژن، جهشهای غیرنول شایعتر هستند (95, 94). بیشتر موارد دیسفیبرینوژنمی به دلیل موتاسیون هتروزیگوت بدمعنی (ندرتاً هموزیگوت یا هتروزیگوت مرکب) در یکی از سه ژن فیبرینوژن ایجاد شده و با الگوی اتوزومال غالب به ارث میرسند. علاوه بر این، موتاسیونهای حذف و یا اضافه شدن[1] یک یا چند نوکلئوتید و تغییر چارچوب نیز در این اختلالات شناسایی شده است (97, 96). بهطورکلی، ناهنجاریهای مولکولی که منجر به اختلال کیفی فیبرینوژن میشوند، بر ساختار و عملکرد مولکول فیبرینوژن شامل برش و آزادسازی فیبرینوپپتید، پلیمریزاسیون فیبرین و تشکیل اتصالات متقاطع فیبرین اثر میگذارند. در این اختلالات، دو موتاسیون بدمعنی (جهشهای نقاط داغ)[2] شایع هستند. موتاسیون در ژن FGA(c.103C>T or c.104G>A)، در اگزون ۲ در اسیدآمینه Arg35 رخ داده و به هیستیدین یا سیستئین تبدیل میشود. این موتاسیون منجر به اتصال ناقص ترومبین و آزاد شدن غیرطبیعی فیبرینوپپتید A میشود (98, 87). اسیدآمینه 301 Arg در اگزون ۸ ژن FGG، ( c.901C>T یا c.902G>A ) نیز به هیستیدین یا سیستئین تغییر یافته که تعاملات D:D را تحت تأثیر قرار داده و متعاقباً منجر به نقص در مراحل اولیه پلیمریزاسیون فیبرین میشود (88, 20). با در نظر گرفتن جهشهای نقاط داغ و اسیدآمینههای مجاور در اگزون ۲ ژن FGA و اگزون ۸ ژن FGG، حدود ۸۵% موارد دیسفیبرینوژنمی قابل تشخیص است، بنابراین، در بررسیهای ژنتیکی ابتدا باید این اگزونها غربالگری شوند (99, 46, 29).
در هیپودیسفیبرینوژنمی، ۳۲ موتاسیون عامل که عمدتاً بدمعنی، بیمعنی و تغییر چارچوب هستند، شناسایی شده است (97, 77). فنوتایپ “هیپو” در هیپودیسفیبرینوژنمی به دلیل سرهمبندی ناقص مولکول فیبرینوژن، کاهش ترشح یا افزایش کلیرانس فیبرینوژن ایجاد میشود، اما فنوتایپ “دیس” اغلب مربوط به پلیمریزاسیون ناقص فیبرین یا اتصال غیرطبیعی کلسیم یا فعالکننده پلاسمینوژن بافتی است (101, 100). چندین مکانیسم مولکولی برای ایجاد این فنوتایپها مطرح شده است؛ از یک طرف، تنها یک موتاسیون میتواند سبب تولید زنجیره فیبرینوژن غیرنرمال شود که ترشح ناکارآمدتری دارد و از طرف دیگر دو موتاسیون هتروزیگوت مجزا میتوانند سبب نقص کیفی و کمیشوند (104-102, 97).
تشخیص آزمایشگاهی اختلالات مادرزادی فیبرینوژن
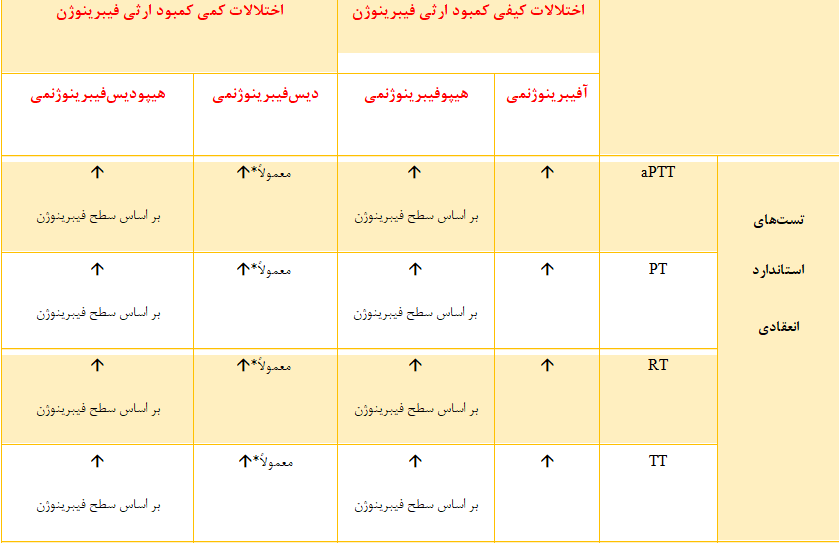
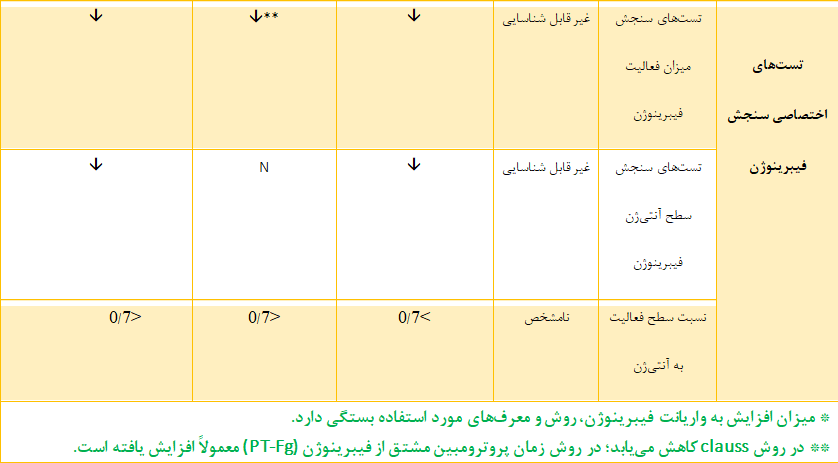
تشخیص آزمایشگاهی اختلالات مادرزادی فیبرینوژن با انجام آزمایشهای روتین انعقادی مثل زمان پروترومبین (PT)[3] و زمان نسبی ترومبوپلاستین فعالشده[4](APTT) آغاز شده و با بررسی سطح فعالیت و آنتیژن فیبرینوژن ادامه مییابد (100). در این اختلالات، تستهای PT و aPTT متناسب با سطح فیبرینوژن طولانی میشوند. علاوه بر این، اگرچه طولانی شدن زمان ترومبین(TT)[5] و زمان رپتیلاز (RT) [6] نیز مشاهده میشود و نشاندهنده کمبود فیبرینوژن هستند، اما از این دو تست بهطور روتین برای تشخیص این اختلالات استفاده نمیشود (جدول ۳-۶) (105). در مرحله بعد، بررسی میزان فعالیت و آنتیژن فیبرینوژن به منظور افتراق بین نقص کیفی یا کمّی کمبود فیبرینوژن، ضروری است (106).
تستهای بررسی فعالیت فیبرینوژن
روش clauss
روش cluass رایجترین روش مورد استفاده برای اندازهگیری سطح فعالیت فیبرینوژن است. در این روش، عملکرد (فعالیت) پلاسمایی فیبرینوژن بر اساس زمان لخته شدن (CT)[7] اندازهگیری میشود (105).
| کادر ۱-۶ اصول انجام روش clauss
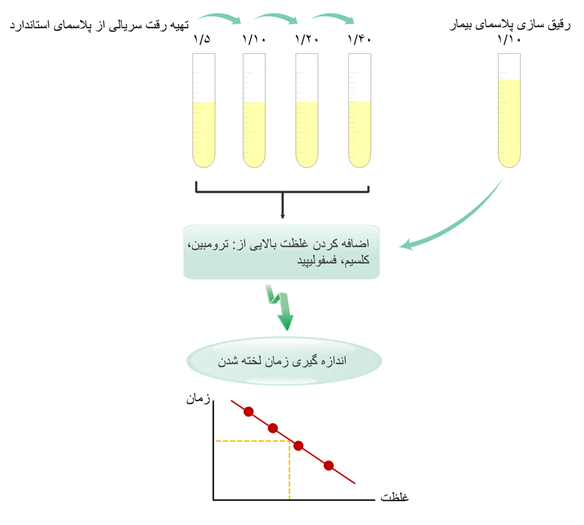
در ابتدا به منظور تهیه منحنی استاندارد، از پلاسمای استاندارد عاری از پلاکت [8](PPP)با غلظت معین فیبرینوژن، رقتهای متوالی (1/40، 1/20، 1/10، 1/5) تهیه میشود. تهیه حداقل سه رقت از پلاسمای استاندارد ضروری است (۱۰۷). هر رقت ابتدا به مدت ۵-۲ دقیقه در ℃۳۷ انکوبه شده و سپس با مقدار مشخصی ترومبین (معمولاً 100 U/ml) مخلوط میگردد (معرفهای تجاری ترومبین در غلظت ۳۵ تا ۲۰۰ واحد در هر میلیلیتر ترومبین انسانی یا گاوی در دسترس هستند) (۱۰۰). در نهایت، برای رسم منحنی استاندارد، زمان انعقاد [9] (CT) بدستآمده از مرحله قبل برای هر رقت، در مقابل غلظتهای مختلف فیبرینوژن (g/L) در یک منحنی لگاریتمی قرار میگیرد. طبق منحنی استاندارد تهیه شده و CT بیمار در رقت 1/10، غلظت فیبرینوژن تعیین میشود (شکل ۴-۶، ۵-۶، ۶-۶). لخته تشکیلشده میتواند با روشهای نوری یا مکانیکی شناسایی شود. روشهای نوری بر اساس تغییر میزان جذب نوری (OD)[10] و روشهای مکانیکی بر اساس استحکام کششی لخته[11] است (107-100). متغیرهای مداخلهگر: مقادیر زیاد قطعات حاصل از تخریب فیبرین (FDPs)[12] و همچنین مهارکنندههای مستقیم ترومبین میتوانند باعث کاهش کاذب میزان فیبرینوژن شوند. علاوه بر این، نمونههای همولیز، لیپمیک و ایکتر نیز میتوانند بر نتایج تست clauss اثر بگذارند. حضور لخته در نمونه، اگرچه به میزان اندک، این تست را مختل میکند (108). تفسیر نتایج تست: فعالیت فیبرینوژن، در آفیبرینوژنمی، غیر قابل شناسایی است اما در هیپوفیبرینوژنمی، دیسفیبرینوژنمی و هیپودیسفیبرینوژنمی کاهش یافته است (جدول ۳-۶) (109). |
|
شکل 6-4 اصول تست کلاوس در این تست رقت متوالی از PPP تهیه خواهد شد و به آن غلظت بالایی از ترومبین اضافه میشود. با اندازه گیری CT، منحنی استاندارد کشیده میشود. رقت 1/10 از پلاسمای بیمار نیز تهیه شده و به همان روش قبلی CT آن اندازهگیری میشود. در نهایت، میزان فیبرینوژن پلاسمای بیمار با استفاده از منحنی استاندارد تعیین میگردد
|
|
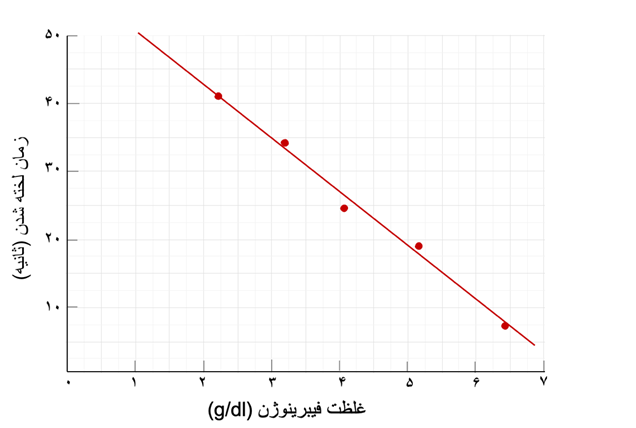
شکل 6-5 نمونهای از منحنی استاندارد در تست کلاوس غلظت فیبرینوژن نمونه بیمار بر اساس این منحنی طبق CT بیمار تعیین میشود |

| شکل 6-6 شکل شماتیک از تست های اندازهگیری فعالیت فیبرینوژن (برگرفته از ۵۱) |
سنجش فیبرینوژن با روش مشتق از تست زمان پروترومبین
سنجش فیبرینوژن با روش مشتق از زمان پروترومبین (PT-Fg)[13] یک روش ساده و مقرون به صرفه است که سطح فعالیت فیبرینوژن را بهطور غیرمستقیم اندازهگیری میکند. اساس این آزمایش تغییر در عبور نوری یا پراکنش از منحنی PT است (شکل ۶-۶).
| کادر ۲-۶. اصول سنجش فیبرینوژن با روش مشتق از تست زمان پروترومبین (PT-Fg)
ابتدا به منظور رسم منحنی استاندارد، از پلاسمای استاندارد با میزان مشخص فیبرینوژن، رقتهای متوالی تهیه میشود. تست PT-Fg برای هر رقت انجام شده و میزان جذب نوری هر غلظت در مقابل سطح فعالیت فیبرینوژن در منحنی قرار میگیرد، سپس میزان PT-Fg بیمار طبق منحنی استاندارد تعیین میشود (۱۱۱). متغیرهای مداخلهگر: نوع معرفها، دستگاه و روش کالیبراسیون بر تست PT-Fg تأثیر بسزایی دارد اما برخلاف تست clauss، مقادیر بالای FDPs بر نتایج این تست تأثیر نمیگذارد. از آنجا که این تست بر اساس اندازهگیری جذب نوری است، نمونههای لیپمیک و ایکتریک میتوانند در نتایج آزمایش اختلال ایجاد کنند. تفسیر: در آفیبرینوژنمی، سطح فعالیت فیبرینوژن در تست PT-Fg غیر قابل شناسایی است، اما در هیپوفیبرینوژنمی کاهش مییابد. در دیسفیبرینوژنمی و هیپودیسفیبرینوژنمی نتایج تست PT-Fg با تست clauss تناقض دارد که در ادامه در بخش مشکلات آزمایشگاهی بحث خواهد شد. |
سنجش انعقادپذیری تام فیبرینوژن[14]
این آزمون که به clottable protein، clot-recovery و روش clottability نیز معروف است، تستی بسیار دقیق بوده و اساس آن تشکیل فیبرین از فیبرینوژن است. اگرچه این تست صحت و تکرارپذیری بالایی دارد و مدتها به عنوان روش مرجع برای سنجش فیبرینوژن مورد استفاده قرار میگرفت، اما به دلیل زمانبر بودن تست و لزوم مهارت برای انجام آن، استفاده از این روش محدود شده است (111, 110).
| کادر ۳-۶ اصول سنجش انعقاد پذیری تام فیبرینوژن
در این تست متعاقب اضافه کردن ترومبین (بدون اضافه کردن +Ca2)، فیبرینوژن لخته میشود. لخته تشکیلشده، شسته شده و در اوره قلیایی یا معرفهای دیگر حل میشود. در نهایت غلظت فیبرینوژن با استفاده از اسپکتروفتومتر (معمولاً در طول موج 282nm) تعیین میشود. ماهیت لخته تقریباً تماماً از فیبرین است، در نتیجه غلظت پروتئین اندازهگیری شده معادل با غلظت فیبرینوژن در نظر گرفته میشود. |
روشهای ایمونولوژیک
سطح آنتیژن فیبرینوژن به وسیله روشهای مختلفی از جمله الیزا [15] (ELISA)، ایمونودیفیوژن شعاعی[16]، ایمونونفلومتری[17] و ایمونوتوربیدومتری[18] تعیین میشود. تکنیک الکتروفورز نیز به عنوان یک روش غربالگری برای شناسایی واریانتهای فیبرینوژن استفاده میشود که اساس آن شناسایی واریانتهای غیرطبیعی فیبرینوژن با توجه به سرعت حرکت باندها است (107). سطح آنتیژن فیبرینوژن در آفیبرینوژنمی غیر قابل شناسایی است اما در هیپوفیبرینوژنمی کاهش مییابد. در دیسفیبرینوژنمی، مقدار آنتیژن فیبرینوژن در محدوده طبیعی قرار دارد اما در هیپودیسفیبرینوژنمی کاهش مییابد (جدول ۳-۶).
جدول ۶.۳ تستهای تشخیصی استاندارد و اختصاصی کمبود مادرزادی فیبرینوژن
سنجشهای جامع هموستاز[19]
گاهی علاوه بر تستهای روتین هموستاز، لازم است ارزیابی جامعتری برای بیماران انجام شود. ترومبوالاستومتری[20]، بهطور گسترده به عنوان تست end-point کمکی در کارآزماییهای بالینی تصادفی بر روی کنسانترههای فیبرینوژن استفاده میشود (112). هنگامی که این تستها روی نمونه پلاسما انجام شود، نتایج این تست بیشتر به محتوای فیبرینوژن و شبکه فیبرینی ایجادشده وابسته است (113)، بنابراین، از این تست می توان در مانیتورینگ اثربخشی تزریق فیبرینوژن استفاده کرد (114). همچنین، این تست به منظور مدیریت سیستم هموستاز در جراحیهای با ریسک بالای خونریزی قابل استفاده است (115). با این وجود، همچنان نقش ترومبوالاستومتری در اختلالات کیفی فیبرینوژن مورد بحث است.
چندین متغیر از ترومبوالاستوگرافی (TEG)[21] با واریانتهای خاصی از فیبرینوژن همبستگی زیادی دارند که این موضوع باعث محدود شدن استفاده از این روش به طیف کوچکی از بیماران مبتلا به اختلالات مادرزادی فیبرینوژن شده است (116). در یک مطالعه که بر روی تعداد کمی از بیماران صورت گرفته است، منحنی TEG در بیماران هیپودیسفیبرینوژنمی منحصر به فرد است، اما با تظاهرات بالینی مرتبط نیست (117). به نظر میرسد که ترومبوالاستومتری چرخش [22] (ROTEM) قادر به افتراق بیماران مبتلا به هیپوفیبرینوژنمی از دیسفیبرینوژنمی و همچنین شناسایی زنان در معرض خطر مشکلات بارداری است (118, 119).
سنجش تولید ترومبین [23] (TGA) بهطور گسترده به منظور بررسی ترومبوز در بیماران مبتلا به آفیبرینوژنمی استفاده میشود. از آنجا که فیبرینوژن به واسطه ترومبین به فیبرین تبدیل میشود، ارتباط بین ترومبین و فیبرین به خوبی شناخته شده است (120, 121). به دلیل اینکه ترومبین بر روی فیبرین پلیمریزهشده جذب میشود، حضور فیبرین یا فیبرینوژن، بر ایجاد ترومبین اثر میگذارد (122). با توجه به نوع آزمایش، حضور فیبرین یا فیبرینوژن، منحنی ایجاد ترومبین را به طرق مختلفی تنظیم میکنند. اگر از فاز مایع در اطراف لخته در حال تشکیل نمونهبرداری کرده و میزان ترومبین آزاد را اندازهگیری کنیم، این میزان در پلاسمایی که فیبرینوژن آن حذف شده است، نسبت به پلاسمای طبیعی بیشتر است (122). در مقابل، در سنجش کالیبره و اتوماتیک ترومبوگرام[24] که میزان ترومبین با ناحیه کاتالیک دستنخورده[25] را اندازه میگیرد، میزان کمتری از ترومبین مشاهده شده است (123,124). هرچند، هنوز اثبات نشده است که آیا TGA میتواند هموستاز را در بیماران مبتلا به اختلالات مادرزادی فیبرینوژن بررسی کند یا خیر (125). در اختلالات کیفی فیبرینوژن، بررسی ساختار لخته فیبرینی، آگاهی ما را نسبت به فنوتیپ بیمار افزایش میدهد. دیسفیبرینوژنمی مرتبط با ترومبوز، عمدتاً به دلیل تغییر استحکام شبکه فیبرینی به دنبال اتصال ضعیف پلاسمینوژن[26] یا فعالکننده پلاسمینوژن بافتی[27] به فیبرینوژن غیرطبیعی و در نهایت کاهش فیبرینولیز ایجاد میشود (127, 126). افزایش نفوذپذیری لخته (منافذ بزرگتر و تراکم کمتر فیبرین) در بیماران مبتلا به دیسفیبرینوژنمی با فنوتیپ خونریزی گزارش شده است ( 128). اگرچه این آزمایشها منحصراً در آزمایشگاههای تحقیقاتی انجام میشوند، اما به تعیین بهتر فنوتیپ بالینی بیمار کمک میکنند.
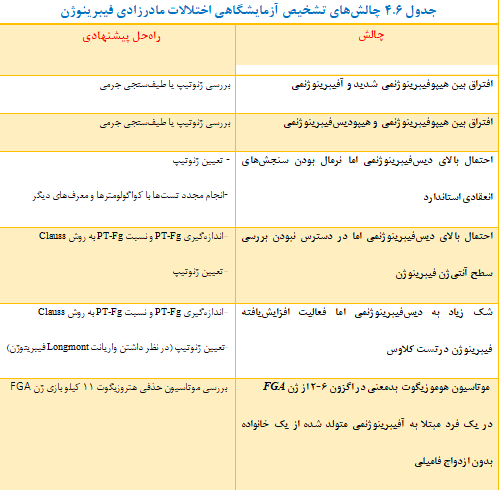
چالشهای تشخیص آزمایشگاهی اختلالات مادرزادی فیبرینوژن
در تشخیص اختلالات مادرزادی فیبرینوژن، چالشهایی وجود دارد که در این قسمت بهطور خاص بررسی خواهند شد (جدول ۴-۶). یکی از این چالشها افتراق هیپوفیبرینوژنمی شدید (gr/l۵/۰> فیبرینوژن) از آفیبرینوژنمی است، چرا که کمترین سطح قابل اندازهگیری فیبرینوژن gr/L۵/۰ است (129). بهطور مشابه، افتراق هیپوفیبرینوژنمی از هیپودیسفیبرینوژنمی نیز چالشبرانگیز است. میزان مقادیر بالاتر از ۷/۰ برای نسبت فعالیت فیبرینوژن به آنتیژن (Activity/Antigen) به عنوان یک ابزار تشخیصی در اختلالات کیفی فیبرینوژن به کار برده میشود (130). البته این cut-off بهطور قطعی مورد قبول نیست و خصوصاً برای تشخیص هیپودیسفیبرینوژنمی حساسیت و اختصاصیت پایینی دارد (77). در این موارد، استفاده از طیفسنجی جرمی[28] کمککننده است، زیرا حتی میتواند مقادیر کم واریانت فیبرینوژن در گردش را شناسایی کند (131). علاوه بر این، روش بیان پروتئین نوترکیب نیز به بررسی ترشح واریانتهای فیبرینوژن کمک میکند (132). از دیگر چالشهای تشخیصی این است که سنجش میزان فعالیت فیبرینوژن در اختلالات کیفی فیبرینوژن به چندین متغیر وابسته است. تست PT-Fg میزان فعالیت فیبرینوژن را حدود ۵ برابر بیشتر از روش clauss نشان میدهد، بنابراین نمیتوان از این تست برای غربالگری اختلالات مادرزادی فیبرینوژن استفاده کرد (47). البته تست PT-Fg، میزان آنتیژن فیبرینوژن را دقیق تعیین میکند و هنگامی که تست دیگری بدین منظور موجود نیست میتواند به عنوان انتخاب دوم استفاده گردد (134, 133). باید این نکته را در نظر داشت که در حضور تعدادی از واریانتهای فیبرینوژن مانند Longmont و Bordeaux، روش clauss میزان فعالیت فیبرینوژن را در مقایسه با PT-Fg بالاتر تخمین میزند (136، 135). بهطور مشابه، تستهای انعقادی استاندارد نیز حساسیت پایینی در تشخیص دیسفیبرینوژنمی دارند و نتایج آنها تحت تأثیر کواگولومتر، معرفها و تشخیص end-point قرار میگیرند (137).
منابع:
- Neerman-Arbez M, De Moerloose P, Casini A, editors. Laboratory and genetic investigation of mutations accounting for congenital fibrinogen disorders. Seminars in thrombosis and hemostasis; 2016: Thieme Medical Publishers.
- Palla R, Peyvandi F, Shapiro ADJB. Rare bleeding disorders: diagnosis and treatment. 2015;125(13):2052-61.
- Casini A, De Moerloose P, Neerman-Arbez M, editors. Clinical features and management of congenital fibrinogen deficiencies. Seminars in thrombosis and hemostasis; 2016: Thieme Medical Publishers.
- Dorgalaleh A, Alavi SER, Tabibian S, Soori S, Moradi Eh, Bamedi T, et al. Diagnosis, clinical manifestations and management of rare bleeding disorders in Iran. 2017;22(4):224-30.
- De Moerloose P, Casini A, Neerman-Arbez M, editors. Congenital fibrinogen disorders: an update. Seminars in thrombosis and hemostasis; 2013: Thieme Medical Publishers.
- Menache DJAotNYAoS. Congenital fibrinogen abnormalities. 1983;408(1):121-30.
- Casini A, Neerman‐Arbez M, Ariens R, De Moerloose PJJoT, Haemostasis. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. 2015;13(6):909-19.
- De Moerloose P, Neerman-Arbez MJEoobt. Treatment of congenital fibrinogen disorders. 2008;8(7):979-92.
- Molmenti EP, Ziambaras T, Perlmutter DJJoBC. Evidence for an acute phase response in human intestinal epithelial cells. 1993;268(19):14116-24.
- Lee SY, Lee KP, Lim JWJT, haemostasis. Identification and biosynthesis of fibrinogen in human uterine cervix carcinoma cells. 1996;75(03):466-70.
- Haidaris PJSJB. Induction of fibrinogen biosynthesis and secretion from cultured pulmonary epithelial cells. 1997;89(3):873-82.
- Soria J, Soria C, Samama M, Poirot E, Kling CJP-b. Human platelet fibrinogen: a protein different from plasma fibrinogen. 1976;24:15-7.
- Francis CW, Nachman RL, Marder VJJT, haemostasis. Plasma and platelet fibrinogen differ in γ chain content. 1984;52(01):084-8.
- Söderqvist T, Blombäck BJN. Fibrinogen structure and evolution. 1971;58(1):16-23.
- Blombäck BJTr. Fibrinogen structure, activation, polymerization and fibrin gel structure. 1994;75(3):327.
- Mosesson MW, editor Fibrinogen structure and fibrin clot assembly. Seminars in thrombosis and hemostasis; 1998: Copyright© 1998 by Thieme Medical Publishers, Inc.
- Fish RJ, Neerman-Arbez MJT, haemostasis. Fibrinogen gene regulation. 2012;108(09):419-26.
- Henry I, Uzan G, Weil D, Nicolas H, Kaplan J, Marguerie C, et al. The genes coding for A alpha-, B beta-, and gamma-chains of fibrinogen map to 4q2. 1984;36(4):760.
- Crabtree GR, Comeau CM, Fowlkes DM, Fornace Jr AJ, Malley JD, Kant JAJJomb. Evolution and structure of the fibrinogen genes: random insertion of introns or selective loss? 1985;185(1):1-19.
- Asselta R, Duga S, Tenchini MLJJoT, Haemostasis. The molecular basis of quantitative fibrinogen disorders. 2006;4(10):2115-29.
- Macrae FL, Domingues MM, Casini A, Ariens RA, editors. The (Patho) physiology of Fibrinogen γ′. Seminars in thrombosis and hemostasis; 2016: Thieme Medical Publishers.
- De Moerloose P, Neerman-Arbez M, editors. Congenital fibrinogen disorders. Seminars in thrombosis and hemostasis; 2009: © Thieme Medical Publishers.
- Neerman-Arbez M, Tirefort Y, de Moerloose PJJOCD. Can mutations identified in congenital fibrinogen disorders explain the clinical manifestations? 2010;2(2).
- Stanciakova L, Kubisz P, Dobrotova M, Stasko JJEroh. Congenital afibrinogenemia: From etiopathogenesis to challenging clinical management. 2016;9(7):639-48.
- Ogata Y, Hepplmann CJ, Charlesworth MC, Madden BJ, Miller MN, Kalli KR, et al. Elevated levels of phosphorylated fibrinogen-α-isoforms and differential expression of other post-translationally modified proteins in the plasma of ovarian cancer patients. 2006;5(12):3318-25.
- Parastatidis I, Thomson L, Burke A, Chernysh I, Nagaswami C, Visser J, et al. Fibrinogen β-chain tyrosine nitration is a prothrombotic risk factor. 2008;283(49):33846-53.
- Medved L, Weisel J, FIBRINOGEN, THROMBOSIS FXSOTSSCOTISO, Thrombosis HJJo, Haemostasis. Recommendations for nomenclature on fibrinogen and fibrin. 2009;7(2):355-9.
- Mosesson MJJoT, Haemostasis. Fibrinogen and fibrin structure and functions. 2005;3(8):1894-904.
- Olexa SA, Budzynski AZJPotNAoS. Evidence for four different polymerization sites involved in human fibrin formation. 1980;77(3):1374-8.
- Gorkun OV, Veklich YI, Medved’ LV, Henschen AH, Weisel JWJB. Role of the. alpha. C Domains of Fibrin in Clot Formation. 1994;33(22):6986-97.
- Li X, Galanakis D, Gabriel DAJJoBC. Transient intermediates in the thrombin activation of fibrinogen evidence for only the desAA species. 1996;271(20):11767-71.
- Dorgalaleh A, Rashidpanah J. Blood coagulation factor XIII and factor XIII defciency. Blood Rev. 2016;30(6):461–75.
- Muszbek L, Adany R, Mikkola HJCricls. Novel aspects of blood coagulation factor XIII. I. Structure, distribution, activation, and function. 1996;33(5):357-421.
- Muszbek L, Yee VC, Hevessy ZJTR. Blood coagulation factor XIII: structure and function. 1999;94(5):271-305.
- Dorgalaleh A, Rashidpanah JJBr. Blood coagulation factor XIII and factor XIII deficiency. 2016;30(6):461-75.
- Drew AF, Liu H, Davidson JM, Daugherty CC, Degen JL. Wound-healing defects in mice lacking fbrinogen. Blood. 2001;97(12):3691–8.
- Clark RA. Fibrin and wound healing. Ann N Y Acad Sci. 2001;936(1):355–67.
- Ikeda Y, Handa M, Kawano K, Kamata T, Murata M, Araki Y, et al. The role of von Willebrand factor and fbrinogen in platelet aggregation under varying shear stress. J Clin Investig. 1991;87(4):1234.
- Staton CA, Brown NJ, Lewis CE. The role of fbrinogen and related fragments in tumour angiogenesis and metastasis. Expert Opin Biol Ther. 2003;3(7):1105–20.
- Rybarczyk BJ, Lawrence SO, Simpson-Haidaris PJ. Matrix-fbrinogen enhances wound closure by increasing both cell proliferation and migration. Blood. 2003;102(12):4035–43.
- Suh TT, Holmbäck K, Jensen NJ, Daugherty CC, Small K, Simon DI, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fbrinogen-defcient mice. Genes Dev. 1995;9(16):2020–33.
- Cheresh DA, Berliner SA, Vicente V, Ruggeri ZM. Recognition of distinct adhesive sites on fbrinogen by related integrins on platelets and endothelial cells. Cell. 1989;58(5):945–53.
- Peyvandi F. Epidemiology and treatment of congenital fbrinogen defciency. Thromb Res. 2012;130:S7–S11.
43. El Boussaadni Y, Benajiba N, El Ouali A, Amrani R, Rkain M. Congenital afbrinogenemia: a case report. Archives de pediatrie: organe offciel de la Societe francaise de. Pediatrie. 2015;22(1):50–2. - Peyvandi F, Palla R, Menegatti M, Siboni S, Halimeh S, Faeser B, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012;10(4):615–21.
45. Peyvandi F, Di Michele D, Bolton-Maggs P, Lee C, Tripodi A, Srivastava A. Classifcation of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity. J Thromb Haemost. 2012;10(9):1938–43.
46. Lebreton A, Casini A. Diagnosis of congenital fbrinogen disorders. Ann Biol Clin. 2016;74:405–12.
47. Cunningham MT, Brandt JT, Laposata M, Olson JD. Laboratory diagnosis of dysfbrinogenemia. Arch Pathol Lab Med. 2002;126(4):499–505.
48. Miesbach W, Schenk J, Alesci S, Lindhoff-Last E. Comparison of the fbrinogen Clauss assay and the fbrinogen PT derived method in patients with dysfbrinogenemia. Thromb Res. 2010;126(6):e428–e33. - Al-Mondhiry H, Ehmann WC. Congenital afbrinogenemia. Am J Hematol. 1994;46(4):343–7.
- Henselmans J, Meijer K, Haaxma R, Hew J, van Der Meer J. Recurrent spontaneous intracerebral hemorrhage in a congenitally afbrinogenemic patient. Stroke. 1999;30(11):2479–82.
- Taslimi R, Golshani K. Thrombotic and hemorrhagic presentation of congenital hypo/afbrinogenemia. Am J Emerg Med. 2011;29(5):573.e3–5.
52. Paolini R, Sartori MT, Fiorin F, Gorinati M, Boeri G, Girolami A. Perinatal intracranial hemorrhage as frst manifestation of congenital hypofbrinogenemia. Clin Appl Thromb Hemost. 1996;2(1):60–3. - Fettah A, Gökçebay DG, Çulha V, Yaralı N, Tunç B, Özbek N. A rare complication of congenital afbrinogenemia: bone cysts. Turk J Haematol. 2017;34(2):183. 54. Ehmann WC, Al-Mondhiry H. Congenital afbrinogenemia and splenic rupture. Am J Med. 1994;96(1):92–4.
- Dorgalaleh A, Naderi M, Shamsizadeh M. Morbidity and mortality in a large number of Iranian patients with severe congenital factor XIII defciency. Ann Hematol. 2016;95(3):451–5.
- Trehan A, Fergusson I. Congenital afbrinogenaemia and successful pregnancy outcome. Case report. Br J Obstet Gynaecol. 1991;98(7):722–4.
57. Zdziarska J, Undas A, Basa J, Iwaniec T, Skotnicki AB, De Moerloose P, et al. Severe bleeding and miscarriages in a hypofbrinogenemic woman heterozygous for the γAla82Gly mutation. Blood Coagul Fibrinolysis. 2009;20(5):374–6. - Santoro C, Massaro F, Venosi S, Capria S, Baldacci E, Foà R, Mazzucconi MG. Severe thrombotic complications in congenital afbrinogenemia: a pathophysiological and management dilemma. Semin Thromb Hemost. 2016;42(5):577–82.
- Rottenstreich A, Lask A, Schliamser L, Zivelin A, Seligsohn U, Kalish Y. Thromboembolic events in patients with severe inherited fbrinogen defciency. J Thromb Thrombolysis. 2016;42(2):261–6.
- Sartori MT, Milan M, de Bon E, Fadin M, Pesavento R, Zanon E. Thrombosis of abdominal aorta in congenital afbrinogenemia: case report and review of literature. Haemophilia. 2015;21(1):88–94.
- Frenkel E, Duksin C, Herman A, Sherman DJ. Congenital hypofbrinogenemia in pregnancy: report of two cases and review of the literature. Obstet Gynecol Surv. 2004;59(11):775–9.
- Awasthy N, Aggarwal K, Gupta H, Saluja S. Congenital hypofbrinogenemia. Indian Pediatr. 2004;41(2):185–6.
63. Hasselback R, Marion RB, Thomas J. Congenital hypofbrinogenemia in fve members of a family. Can Med Assoc J. 1963;88(1):19.
64. Bay A, Coskun E, Leblebisatan G, Sivasli E. Epidural hematoma and cephalohematoma with congenital hypofbrinogenemia. Blood Coagul Fibrinolysis. 2012;23(3):229–31. - Rubbia-Brandt L, Neerman-Arbez M, Rougemont A-L, Malé P-J, Spahr L. Fibrinogen gamma375 arg→ trp mutation (fbrinogen aguadilla) causes hereditary hypofbrinogenemia, hepatic endoplasmic reticulum storage disease and cirrhosis. Am J Surg Pathol. 2006;30(7):906–11.
- Wehinger H, Klinge O, Alexandrakis E, Schürmann J, Witt J, Seydewitz H. Hereditary hypofbrinogenemia with fbrinogen storage in the liver. Eur J Pediatr. 1983;141(2):109–12.
- Pfeifer U, Ormanns W, Klinge O. Hepatocellular fbrinogen storage in familial hypofbrinogenemia. Virchows Arch B. 1981;36(1):247–55.
68. Casini A, Sokollik C, Lukowski S, Lurz E, Rieubland C, Moerloose P, et al. Hypofbrinogenemia and liver disease: a new case of Aguadilla fbrinogen and review of the literature. Haemophilia. 2015;21(6):820–7. - Asselta R, Robusto M, Braidotti P, Peyvandi F, Nastasio S, D’antiga L, et al. Hepatic fbrinogen storage disease: identifcation of two novel mutations (p. Asp316Asn, fbrinogen Pisa and p. Gly366Ser, fbrinogen Beograd) impacting on the fbrinogen γ-module. J Thromb Haemost. 2015;13(8):1459–67.
- Puls F, Goldschmidt I, Bantel H, Agne C, Bröcker V, Dämmrich M, et al. Autophagyenhancing drug carbamazepine diminishes hepatocellular death in fbrinogen storage disease. J Hepatol. 2013;59(3):626–30.
- Casini A, Duval C, Pan X, Tintillier V, Biron-Andreani C, Ariëns R. Fibrin clot structure in patients with congenital dysfbrinogenaemia. Thromb Res. 2016;137:189–95.
- Casini A, Blondon M, Lebreton A, Koegel J, Tintillier V, de Maistre E, et al. Natural history of patients with congenital dysfbrinogenemia. Blood. 2015;125(3):553–61.
- Miesbach W, Galanakis D, Scharrer I. Treatment of patients with dysfbrinogenemia and a history of abortions during pregnancy. Blood Coagul Fibrinolysis. 2009;20(5):366–70.
- Miesbach W, Scharrer I, Henschen A, Neerman-Arbez M, Spitzer S, Galanakis D. Inherited dysfbrinogenemia: clinical phenotypes associated with fve different fbrinogen structure defects. Blood Coagul Fibrinolysis. 2010;21(1):35–40.
- Hayes T. Dysfbrinogenemia and thrombosis. Arch Pathol Lab Med. 2002;126(11):1387–90.
- Rowczenio D, Stensland M, de Souza GA, Strøm EH, Gilbertson JA, Taylor G, et al. Renal amyloidosis associated with fve novel variants in the fbrinogen A alpha chain protein. Kidney Int Rep. 2017;2:461–9.
- Casini A, Brungs T, Lavenu-Bombled C, Vilar R, Neerman-Arbez M, Moerloose P. Genetics, diagnosis and clinical features of congenital hypodysfbrinogenemia: a systematic literature review and report of a novel mutation. J Thromb Haemost. 2017;15(5):876–88.
- Deering SH, Landy HJ, Tchabo N, Kessler C. Hypodysfbrinogenemia during pregnancy, labor, and delivery. Obstet Gynecol. 2003;101(5, Part 2):1092–4.
- Cheah CY, Brennan SO, Kennedy H, Januszewicz EH, Maxwell E, Burbury K. Fibrinogen Melbourne: a novel congenital hypodysfbrinogenemia caused by γ326Cys-Phe in the fbrinogen γ chain, presenting as massive splanchnic venous thrombosis. Blood Coagul Fibrinolysis. 2012;23(6):563–5.
- Ebert RF, Bell WR. Fibrinogen Baltimore II: congenital hypodysfbrinogenemia with delayed release of fbrinopeptide B and decreased rate of fbrinogen synthesis. Proc Natl Acad Sci. 1983;80(23):7318–22.
- Lefebvre P, Velasco PT, Dear A, Lounes KC, Lord ST, Brennan SO, et al. Severe hypodysfbrinogenemia in compound eterozygotes of the fbrinogen AαIVS4+ 1G> T mutation and an AαGln328 truncation (fbrinogen Keokuk). Blood. 2004;103(7):2571–6.
82. Jayo A, Arnold E, González-Manchón C, Green D, Lord ST. Hypodysfbrinogenemia causing mild bleeding and thrombotic complications in a compound heterozygote of AαIVS4+ 1G> T mutation and Aα4841delC truncation (AαPerth). Thromb Haemost. 2009;101(4):770. - Duga S, Asselta R, Santagostino E, Zeinali S, Simonic T, Malcovati M, et al. Missense mutations in the human β fbrinogen gene cause congenital afbrinogenemia by impairing fbrinogen secretion. Blood. 2000;95(4):1336–41. 84. Neerman-Arbez M, De Moerloose P, Bridel C, Honsberger A, Schönbörner A, Rossier C, et al. Mutations in the fbrinogen Aα gene account for the majority of cases of congenital afbrinogenemia. Blood. 2000;96(1):149–52.
- Neerman-Arbez M. The molecular basis of inherited afbrinogenaemia. Thromb Haemost. 2001;86(1):154–63.
86. Uzan G, Courtois G, Besmond C, Frain M, Sala-Trepat J, Kahn A, et al. Analysis of fbrinogen genes in patients with congenital afbrinogenemia. Biochem Biophys Res Commun. 1984;120(2):376–83. - Neerman-Arbez M, De Moerloose P. Mutations in the fbrinogen gene cluster accounting for congenital afbrinogenemia: an update and report of 10 novel mutations. Hum Mutat. 2007;28(6):540.
- Brennan SO, Fellowes AP, George PM. Molecular mechanisms of hypo-and afbrinogenemia. Ann N Y Acad Sci. 2001;936(1):91–100.
89. Neerman-Arbez M, De Moerloose P, Honsberger A, Parlier G, Arnuti B, Biron C, et al. Molecular analysis of the fbrinogen gene cluster in 16 patients with congenital afbrinogenemia: novel truncating mutations in the FGA and FGG genes. Hum Genet. 2001;108(3):237–40. - Neerman-Arbez M, Vu D, Abu-Libdeh B, Bouchardy I, Morris MA. Prenatal diagnosis for congenital afbrinogenemia caused by a novel nonsense mutation in the FGB gene in a Palestinian family. Blood. 2003;101(9):3492–4.
- Asselta R, Spena S, Duga S, Peyvandi F, Malcovati M, Mannucci PM, et al. Analysis of Iranian patients allowed the identifcation of the frst truncating mutation in the fbrinogen Bbeta-chain gene causing afbrinogenemia. Haematologica. 2002;87(8):855–9.
92. Brennan SO, Davis RL, Conard K, Savo A, Furuya KN. Novel fbrinogen mutation γ314Thr→ Pro (fbrinogen AI duPont) associated with hepatic fbrinogen storage disease and hypofbrinogenaemia. Liver Int. 2010;30(10):1541–7. - Lee MJ, Venick R, Bhuta S, Li X, Wang HL. Hepatic fbrinogen storage disease in a patient with hypofbrinogenemia: report of a case with a missense mutation of the FGA gene. Semin Liver Dis. 2015;35:439–43.
- Asselta R, Platè M, Robusto M, Borhany M, Guella I, Soldà G, et al. Clinical and molecular characterisation of 21 patients affected by quantitative fbrinogen defciency. Thromb Haemost. 2015;113(3):567–76.
- Sumitha E, Jayandharan G, Arora N, Abraham A, David S, Devi G, et al. Molecular basis of quantitative fbrinogen disorders in 27 patients from India. Haemophilia. 2013;19(4):611–8.
- Koopman J, Haverkate F, Grimbergen J, Egbring R, Lord S. Fibrinogen Marburg: a homozygous case of dysfbrinogenemia, lacking amino acids A alpha 461-610 (Lys 461 AAA–> stop TAA). Blood. 1992;80(8):1972–9.
- Vu D, Neerman-Arbez M. Molecular mechanisms accounting for fbrinogen defciency: from large deletions to intracellular retention of misfolded proteins. J Thromb Haemost. 2007;5(s1):125–31.
- Zhou J, Ding Q, Chen Y, Ouyang Q, Jiang L, Dai J, et al. Clinical features and molecular basis of 102 Chinese patients with congenital dysfbrinogenemia. Blood Cell Mol Dis. 2015;55(4):308–15.
- Hill M, Dolan G. Diagnosis, clinical features and molecular assessment of the dysfbrinogenaemias. Haemophilia. 2008;14(5):889–97.
100. Verhovsek M, Moffat KA, Hayward CP. Laboratory testing for fbrinogen abnormalities. Am J Hematol. 2008;83(12):928–31.
101. Sadeghian MH, Keramati MR, Badiei Z, Ravarian M, Ayatollahi H, Rafatpanah H, et al. Alloimmunization among transfusion-dependent thalassemia patients. Asian J Transfus Sci. 2009;3(2):95. - Ridgway HJ, Brennan SO, Faed JM, George PM. Fibrinogen Otago: a major α chain truncation associated with severe hypofbrinogenaemia and recurrent miscarriage. Br J Haematol. 1997;98(3):632–9.
- Martinez J, Holburn R, Shapiro S, Erslev A. Fibrinogen Philadelphia. A hereditary hypodysfbrinogenemia characterized by fbrinogen hypercatabolism. J Clin Investig. 1974;53(2):600.
- Mukai S, Nagata K, Ikeda M, Arai S, Sugano M, Honda T, et al. Genetic analyses of novel compound heterozygous hypodysfbrinogenemia, Tsukuba I: FGG c. 1129+ 62_65 del AATA and FGG c. 1299+ 4 del A. Thromb Res. 2016;148:111–7.
105. Harr JN, Moore EE, Ghasabyan A, Chin TL, Sauaia A, Banerjee A, et al. Functional fbrinogen assay indicates that fbrinogen is critical in correcting abnormal clot strength following trauma. Shock. 2013;39(1):45. - Mackie I, Lawrie A, Kitchen S, Gaffney P, Howarth D, Lowe G, et al. A performance evaluation of commercial fbrinogen reference preparations and assays for Clauss and PT-derived fbrinogen. Thromb Haemost. 2002;87(6):997–1005.
- Mackie IJ, Kitchen S, Machin SJ, Lowe G. Guidelines on fbrinogen assays. Br J Haematol. 2003;121(3):396–404.
- Exner T, Burridge J, Power P, Rickard KA. An evaluation of currently available methods for plasma fbrinogen. Am J Clin Pathol. 1979;71(5):521–7.
- Desvignes P, Bonnet P. Direct determination of plasma fbrinogen levels by heat precipitation. A comparison of the technique against thrombin clottable fbrinogen with spectrophotometry and radial immuno-diffusion. Clin Chim Acta. 1981;110(1):9–17.
110. Besser MW, MacDonald SG. Acquired hypofbrinogenemia: current perspectives. J Blood Med. 2016;7:217. - Casini A, de Moerloose P. Can the phenotype of inherited fbrinogen disorders be predicted? Haemophilia. 2016;22(5):667–75.
- Shapiro SE, Phillips E, Manning RA, Morse CV, Murden SL, Laffan MA, et al. Clinical phenotype, laboratory features and genotype of 35 patients with heritable dysfbrinogenaemia. Br J Haematol. 2013;160(2):220–7.
- Godal H, Brosstad F, Kierulf P. Three new cases of an inborn qualitative fbrinogen defect (Fibrinogen Oslo II). Eur J Haematol. 1978;20(1):57–62.
- Acharya S, Coughlin A, Dimichele DM. Rare Bleeding Disorder Registry: defciencies of factors II, V, VII, X, XIII, fbrinogen and dysfbrinogenemias. J Thromb Haemost. 2004;2(2):248–56.
- Casini A, de Moerloose P, Congenital Fibrinogen Disorders Group. Management of congenital quantitative fbrinogen disorders: a Delphi consensus. Haemophilia. 2016;22(6):898–905.
- Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, et al. Guideline for the diagnosis and management of the rare coagulation disorders. Br J Haematol. 2014;167(3):304–26.
- Bolton-Maggs PH, Perry DJ, Chalmers EA, Parapia LA, Wilde JT, Williams MD, Collins PW, Kitchen S, Dolan G, Mumford AD. The rare coagulation disorders – review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004;10(5):593–628.
[1] Deletion- insertion mutations
[2] Hot spot mutations
[3] Prothrombin time
[4] Activated partial thromboplastin time
[5] Thrombin time
[6] Reptilase time
[7] Clotting time
[8] Platelet poor plasma
[9] Clotting time
[10] Optical density
[11] Tensile clot strength
[12] Fibrin degradation products
[13] Prothrombin time derived fibrinogen
[14] Total clottable fibrinogen assay
[15] Enzyme-linked immunosorbent assay
[16] Radial immunodiffusion
[17] Immunonephelometry
[18] Immunoturbidimetry
[19] Global hemostasis assays
[20] Thromboelastometry
[21] Thromboelastography
[22] Rotational thromboelastography
[23] Thrombin generation assay
[24] Calibrated automated thrombogram assays
[25] Intact catalytic site
[26] Plasminogen
[27] Tissue-type plasminogen activator
[28] Mass spectrometry
تشخیص آزمایشگاهی و درمان اختلالات مادرزادی فیبرینوژن (2)
تشخیص آزمایشگاهی و درمان اختلالات مادرزادی فیبرینوژن
microRNAها در هموستاز و ترومبوز
پاتوفیزیولوژی و یافتههای آزمایشگاهی کواگولوپاتی ناشی از کووید-19
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام
ممنون از زحمات شما