
پیشبینی شدت کمخونی در فرزندان ناقلین
ژن تالاسمی و هموگلوبینوپاتیها
دکتر حبیباله گلافشان
دکتر ناهید نصیری، دکترای تخصصی هماتولوژی
سندرمهای تالاسمی و هموگلوبینوپاتیها گروهی گسترده از اختلالات ارثی سنتز کمی و کیفی هموگلوبین میباشند. این گروه از بیماریها شایعترین اختلالات تکژنی در دنیا بوده که مشکلات جدی سلامت را ببار میآورند. در حال حاضر پیوند سلولهای مادر خونساز تنها راه درمان کمخونیهای شدید است و قبل از آن از درمانهای حمایتی استفاده میشود. زندگی وابسته به تزریق خون نهتنها مشکلات فراوان برای بیمار دربرداشته، بلکه زندگی خانواده را نیز تحت تأثیر قرار میدهد. بهترین راه جلوگیری از تولد فرزند مبتلا به کمخونی شدید، شناسایی ناقلین در سن بلوغ یا قبل از ازدواج و یا در حین بارداری و آموزش صحیح به ناقلین پرخطر کمخونی میباشد.
جدول زیر نامطلوبترین حالت در فرزندان ناقلین پرخطر را نشان میدهد و این به مفهوم آن نیست که تمام فرزندان مبتلا خواهند شد، بلکه در اکثر موارد بدترین حالتی که احتمالاً در 25% فرزندان رخ میدهد را نشان میدهد. در جدول، ناحیه سفید به مفهوم بدون ریسک شناخته شده، رنگ قرمز بیانگر ریسک جدی و کمخونی شدید و رنگ بنفش با خطر جدی کمتر یا کمخونی ملایمتر و رنگ زرد با احتمال خطر نهفته را بیان میکند.
پیشبینی احتمال ریسک کمخونیهای شدید تا متوسط در فرزندان ناقلین ژنهای تالاسمی و هموگلوبینوپاتیها
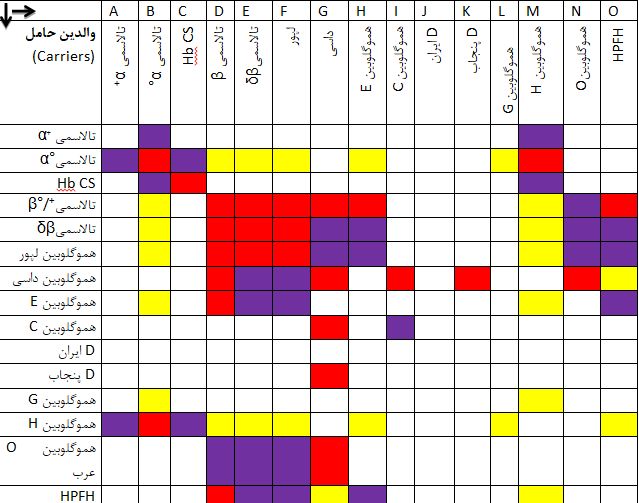
HbCS: Hb Constant Spring
HPFH: Hereditary persistant of fetal hemoglobin
رنگ سفید علامت بدون ریسک شناخته شده (Not known risk)
رنگ قرمز با ریسک جدی و کمخونی شدید (Possible serious risk)
رنگ بنفش با ریسک جدی کمتر (Less serious risk)
رنگ زرد با احتمال ریسک نهفته (Possible hidden risk)
چند مثال:
مثال اول: چنانچه یکی از والدین ناقل تالاسمی α°(αα/–) و دیگری هم ناقل تالاسمی α° (αα/–) باشند در منطقه قرمز جدول تلاقی میکنند زیرا در این حالت احتمال تولد نوزاد مرده مبتلا به هیدروپس فتالیس با هموگلوبین بارت یا سقط در اوایل حاملگی به علت فقدان هر چهار ژن آلفا وجود دارد.
مثال دوم: چنانچه یکی از والدین هتروزیگوت داسی و دیگری هتروزیگوت برای هموگلوبین D پنجاب یا لسآنجلس باشند، باز هم در منطقه قرمز تلاقی میکنند زیرا هتروزیگوت ترکیبی داسی و D پنجاب شبیه آنمی داسی شکل است ولی منطقه تلاقی آن با D ایران در نقطه سفید یا بیخطر است.
مثال سوم: چنانچه هر دو والدین ناقل هموگلوبین ِِD باشند، نقطه تلاقی در منطقه سفید رنگ است زیرا هموزیگوت D ایران و پنجاب کمخونی نمیدهند.
مثال چهارم: حاصل ازدواج فردی با هتروزیگوت HPFH (سنتز مداوم هموگلوبین F) با کمخونی داسی قابل پیشبینی نیست چون ممکن است با سنتز 30 درصدی هموگلوبین F شبیه هتروزیگوت داسی بدون علامت باشد و یا با سنتز کمتر Hb F با آنمی داسی و علائم خفیف و غیرقابل پیشبینی جلوه کند.
تشخیص برخی از ناقلین با آزمایشهای روزمره آزمایشگاهی امکانپذیر است از قبیل افزایش Hb A2 که در نمای میکروسیت هایپوکروم بیانگر تالاسمی ماینور بتاست یا مشاهده مرفولوژی توپ گلف یا اچبادی در رنگآمیزی حیاتی که هموگلوبین اچ را مطرح میکند و یا مشاهده باند A2 بیشتر از 8% که بیانگر حضور هموگلوبینوپاتی و نیاز به الکتروفورز را مطرح میکند.
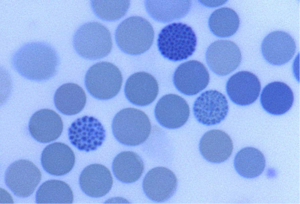
اجسام H در رنگآمیزی حیاتی بهصورت نقطهنقطهای (گلف بادی) در سلولهای قرمز دیده میشوند. اجسام H رسوب تترامرهای β4 بر روی غشای گلبولهای قرمز هستند که به غشا آسیب رسانده و منجر به همولیز میشوند
از سالها پیش تاکنون فرمولهایی جهت افتراق آنمی فقرآهن از سندرمهای تالاسمی ماینور آلفا و بتا ابداع شده است که در جدول زیر مشاهده مینمایید.
تشخیص افتراقی کمخونی فقر آهن از تالاسمی
الگوریتم تشخیصی تالاسمی از آنمی فقر آهن
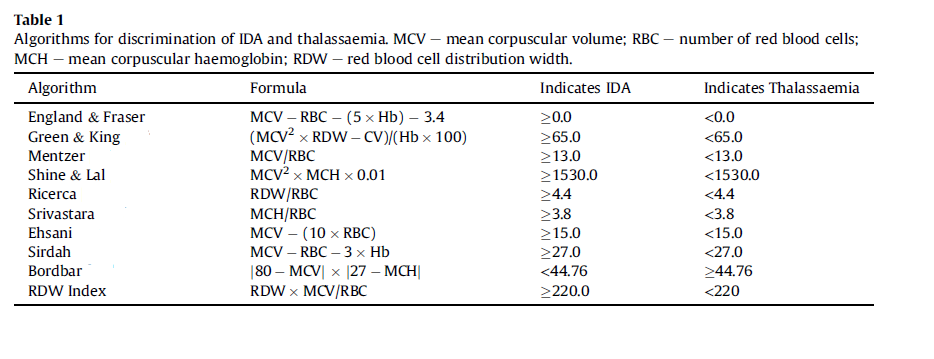
تمام این فرمولها جنبه آکادمیک داشته و بایستی با روشهای الکتروفورز تأئید گردند.
چنانچه روشهای الکتروفورز قانعکننده نباشد بایستی از روشهای مولکولی مانند محاسبه نسبت سنتز زنجیره آلفا به بتا و یا از روشهای مولکولی دیگر مانند Gap-PCR ,MLPA ,ARMS ,ASO ,RE-PCR و Sequencing برای تشخیص صحیح بهره گرفت.
PCR: Polymerase Chain Reaction
MLPA: Multiplex Ligation dependent Probe Amplification
RE: Restriction Enzyme
ASO: Allele Specific Oligonucleotide
ARMS: Amplification Refractory Mutation System
جهشهای تالاسمی بتا ممکن است با فقدان فرآورده زنجیره بتا (°β) همراه باشند یا موجب کاهش تولید زنجیره بتا (+β ) گردند. از مثالهای °β میتوان به جهـــــــــــــــــــــش در رمز اسیدآمینه شماره β°39 و IVS 1-1 ,IVS 2-1 اشاره کرد. جهشهای +β ممکن است مانند جهش IVS 1-110 بسیار شدید و یا مانند 6IVS 2-844 ,IVS 2-745 ,IVS 1- متوسط و یا خفیف (87-, 88-) و یا خاموش (101-) باشند. در حامل خاموش اندکسها و هموگلوبین A2 طبیعی است؛ بنابراین وراثت +β+β ممکن است با تالاسمی اینترمدیای خفیف یا شدید جلوه کند. موارد شدید جهش +β شبیه ° β میباشد و حدود 5 درصد ژن سالم فرآورده دارد. مواردی از +β°β خفیف ممکن است بهصورت اینترمدیا درآید و مواردی از +β+β ممکن است بهصورت تالاسی ماینور بتا جلوه کند؛ بنابراین جلوه بالینی بستگی به نوع جهش β داردکه با روشهای مولکولی قابل تشخیص است.
پیشبینی شدت خطر در ناقلین تالاسمی و هموگلوبینوپاتیها
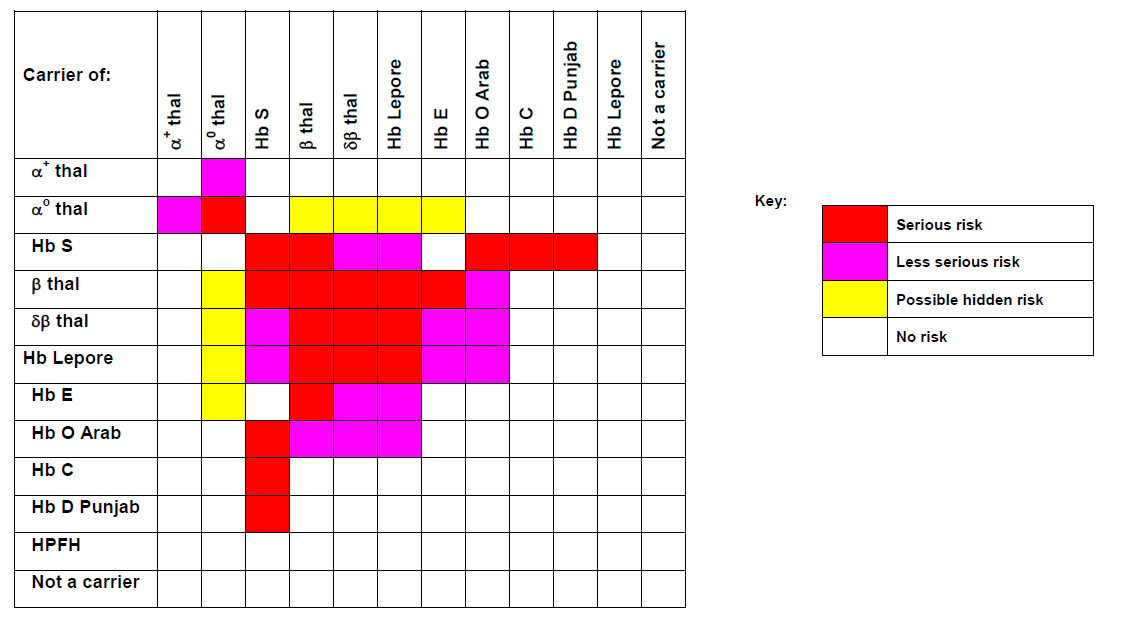
توضیحات تکمیلی در مواردی که هر دو والدین ژنوتایپ تالاسمی +α دارند
تالاسمی +α بهصورت هتروزیگوت یا حامل خاموش (α/αα-) و یا بهصورت هموزیگوت (α-/α-) در همراهی با هموگلوبینوپاتیهای دیگر بیخطر است. موارد استثنا در همراهی با تالاسمی α° (–/αα) است که احتمال فرزند مبتلا به بیماری هموگلوبین اچ با ژنوتایپ –/α- و با کمخونی متوسط شبیه تالاسمی اینترمدیا وجود دارد، بعلاوه در همراهی با هموگلوبین اچ با فقدان سه ژن آلفا نیز احتمال بیماری هموگلوبین اچ در فرزندان میباشد. شایعترین ژنوتایپهای +α در ایران α3.7 وα4.2 است که به ترتیب ادغام α2α1 در α3.7 و حذف 2α در α4.2 مشاهده میشود.
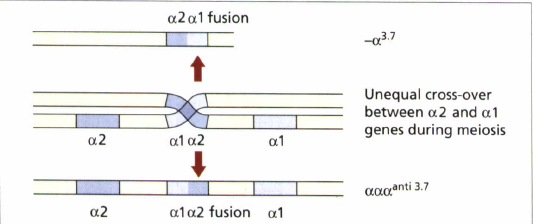
کراسینگاور نابرابر هاپلوتایپهای کروموزوم 16 در ناحیهی ژنهای آلفا در هنگام میوز موجب شکلگیری هاپلوتایپ α3.7 – و هاپلوتایپ سه آلفایی (ααα) میشود
توضیحات تکمیلی در مواردی که هر دو والدین ژنوتایپ تالاسمی (°α یا aa/—) دارند
بدترین حالت و خطر جدی در این حالت ابتلای 25 درصدی جنین به هیدروپس فتالیس با هموگلوبین بارت میباشد که هر 4 ژن آلفا حذف شدهاند (–/–). در این حالت چنانچه ژن زتا هم بهصورت هموزیگوت حذف شده باشد مرگ جنین در ماههای اول و چنانچه ژن زتا حذف نشده باشد تنها با زندگی داخل رحمی سازگار بوده و جنین ورمکرده و مرده به دنیا آمده یا ساعاتی پس از تولد فوت میشود. از شایعترین هاپلوتایپهایα° میتوان به هاپلوتایپهای MED ,SEA و α20.5 با حذف Cis α و هاپلوتایپهای Fil و Thai با حذف Cis α و حذف ژن زتا اشاره کرد.αG یکی از واریانهای آلفاست و در اکثر موارد با حذف α کناری همراهی دارد. در این حالت در همراهی با °α امکان تولد فرزندی با هموگلوبین اچ وجود دارد. امکان دارد گاهی تالاسمی α° در همراهی با هتروزیگوت β°/β بهصورت تالاسمی اینترمدیا جلوه کند.
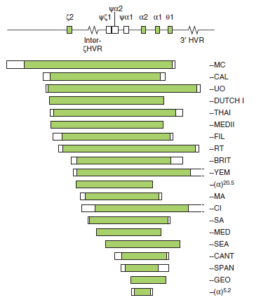
هاپلوتایپهای شایع حذفی سیس آلفا (α0) که در آن، هر دو ژن آلفای کنار هم حذف شدهاند حذفهای MED (مدیترانهای) و SEA (جنوب شرقی آسیا) شایعترین حذفها هستند. رنگ سبز نقاط حذفشده را نشان میدهد
الگوی وراثت تالاسمیهای α و β
همراهی تالاسمی °α با هتروزیگوت کنستانت اسپرینگ (ααCS/αα) CS ممکن است بهصورت بیماری هموگلوبین اچ در فرزندان درآید و در همراهی هموگلوبین اچ با تالاسمی °α احتمال بیماری هموگلوبین اچ و یا هیدروپس فتالیس در فرزندان وجود دارد.

نوزاد مبتلا به هیدروپس فتالیس
در همراهی تالاسمی °α با هاپلوتایپ HPFH حذفی، امکان تولد فرزندی با تالاسمی شبیه ماینور آلفا با افزایش هموگلوبین F و مقدار متغیر A و A2 میباشد. چنانچه زنجیره γ در نوع حذفی جبران کامل سنتز ژن بتا داشته باشد، کمخونی وجود نداشته و گلبولهای قرمز نرموسیت نرموکروم میگردند ولی در نوع غیرحذفی بسته به جهش پروموتر ژن γ، میزان Hb F متغیر است. کمخونی خفیف در حضور یک ژن بتای سالم مشاهده شده و درصورتیکه جهش ژن β در هاپلوتایپ دیگر وجود داشته باشد β°/+ /HPFH میتواند ایجاد کمخونی خفیف تا متوسط در همراهی با تالاسمی °α کند.
نکتههای مهم
- تالاسمی ماینور آلفا با ژنوتایپهای ترانس (α/-α-) و سیس (aa/–) با آزمایشهای CBC و اندازهگیری Hb A2 قابل افتراق نمیباشند و نیازمند آزمایشهای مولکولار مانند Gap PCR برای تشخیص است. حذف سیس آلفا در همراهی با تالاسمیهای آلفا عوارضی مانند بیماری هموگلوبین اچ و هیدروپس فتالیس با هموگلوبین بارت را بدنبال دارد.
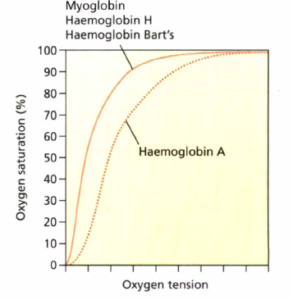
هموگلوبینهای بارت و H میل ترکیبی بسیاری با اکسیژن دارند و در پسدهی اکسیژن فاقد کارایی هستند
- افزایش هموگلوبین A2 (بیشتر از 3/5%) بدون توجه به اندکسهای خون بیانگر تالاسمی ماینور بتاست. گرچه مواردی از قبیل پرکاری تیروئید، کمخونی مگالوبلاستیک، درمان HIV و هموگلوبینهای ناپایدار ممکن است Hb A2 را اندکی افزایش دهند، ولی همیشه افزایش Hb A2 جدی است.
- کاهش اندکسهای گلبول قرمز در تالاسمی ماینورآلفا، ماینور بتا و بیماری هموگلوبین E همراه با افزایش شمارش گلبول قرمز دیده میشود. در مناطقی که شیوع تالاسمی آلفا کم است از کاهش اندکسها همراه با اریتروسیتوز میتوان برای اسکرین تالاسمی ماینور بتا استفاده کرد.
- همراهی وراثت تالاسمی ماینور بتا با تالاسمی آلفا (α/αα-) و یا بندرت (α/-α-) ممکن است به علت بالانس بهتر زنجیرهها، اندکسها را نرمال کند و تشخیص تالاسمی ماینور بتا را مشکل نماید ولی افزایش هموگلوبین A2 این وراثت همزمانی را همراهی میکند.
- یکی از مشکلات تشخیصی تالاسمی ماینور بتا مقدار نرمال یا لب مرز هموگلوبین A2 و حتی گاهی با اندکسهای طبیعی است (حامل خاموش)، گرچه اکثر این موارد تشخیص داده نمیشوند و در همراهی با تالاسمی ماینور بتا ممکن است تالاسمی اینترمدیا را به دنبال داشته باشد. برخی از جهـــــــــــــــــــشها از قبیل92- (C>T) ,101 (C>T)- بهصورت حامل خاموش ماینور بتا خود را نشان میدهند.
- گاهی با وجود اندکسهای تالاسمیک مقدار Hb A2 نرمال یا حتی کمتر از نرمال است؛ برای مثال همراه شدن تالاسمی ماینور بتا با جهشهای ژن دلتا بهصورت سیس و ترانس میتواند موجب کاهش 50 درصدی A2 یا حتی مقدار A2 در حد صفر گردد. در این موارد آزمایشهای مولکولی جهت افتراق از تالاسمی آلفا لازم است.
- دو گونه تالاسمی ماینور بتا با افزایش 4 تا 18 درصدی هموگلوبینF و سطح نرمال A2 همراه است؛ تالاسمی دلتا- بتا با حذف یا جهش ناکارآمد در دو ژن دلتا و بتا و بیان بیشتر ژن گاما که به آن تالاسمی F هم گفته میشود و دیگری HPFH حذفی است که به مفهوم تداوم سنتز هموگلوبین F است که هموگلوبین F بین 3 تا 30 درصد قرار میگیرد.
حذفی به مفهوم حذف شدن ژنهای بتا و دلتا و فعال شدن بیشتر ژن گاما است. در تالاسمی دلتا- بتا اندکسها تالاسمیک، ولی در HPFH نرمال یا در مرز نرمال است.
توضیحات تکمیلی در وراثت ژن هموگلوبین کنستانت اسپرینگ با ژنهای تالاسمی و هموگلوبینوپاتیها
وراثت هاپلوتایپααCS با تالاسمی (aa/–) °α میتواند منجر به بیماری هموگلوبین اچ با کمخونی شدید تا متوسط گردد. هموگلوبین کنستانت اسپرینگ دارای جهش در رمز خاتمه زنجیره آلفا بوده که دارای 31 اسیدآمینه اضافی است. فرم هموزیگوت کنستانت اسپرینگααCS/ααCS با کمخونی شدید و ژاندیس و بزرگی طحال همراه است. گفتنی است که اندکسهای خون در این حالت در محدوده نرمال است.
در همراهی هموزیگوت هموگلوبین کنستانت اسپرینگααCS/ααCS با هاپلوتایپهای هموزیگوت تالاسمی دلتا بتا یا لپور امکان کمخونی شدید تا متوسط نهفته شبیه تالاسمی اینترمدیا وجود دارد ولی با هتروزیگوتهای تالاسمی β، لپور و دلتا-بتا کمخونی نمیدهد.
توضیحات تکمیلی در پیشآمد تولد فرزندی با هاپلوتایپهای تالاسمی β با ژنهای دیگر تالاسمی و هموگلوبینوپاتیها
در هاپلوتایپ °β تولید ژن بتا صفر است و در هاپلوتایپ +β تولید ژن بتا بین 5 تا 30% نرمال است. در حالتهای β°/(Lepore) ,β°/(δβ)°,β°/β و β°/βE آنمی کولیز با کمخونی شدید ظهور میکند ولی ترکیب هاپلوتایپهای +β+β با تالاسمی اینترمدیا همراه است. در این حالت الگوی الکتروفورز FAA2 است در حالی که در β°β° الگو بهصورت FA2 است.
توجه داشته باشید که وقتی هموگلوبینهای D یا O یا S و یا E در شخصی بیشتر از 90% است تنها به هموزیگوتهای DD یا OO یا SS یا EE فکر نکنید بلکه با توجه به کاهش MCV و MCH ممکن است هتروزیگوتهای ترکیبی هموگلوبینهای فوق با تالاسمی باشد که در ازدواج با تالاسمی ماینور بتا امکان تولد فرزندی مبتلا به تالاسمی ماژور وجود دارد؛ برای مثال از ازدواج شخصی با Hb=12 و Hb A2=5% و MCV=64 با شخصی با HbD=95% و Hb A2=4% و HbF=1%، فوراً تصور هموزیگوت D نداشته باشید. در مثال فوق احتمال هتروزیگوت ترکیبی β°/βD زیاد است که در ازدواج با β°/β حاصل آن میتواند °β°β یا آنمی کولیز باشد ولی از ازدواج ناقل تالاسمی با هتروزیگوت (AD) D یا هموزیگوت (DD) D مشکلی ایجاد نمیشود.
ترکیب β°/βE به علت ناپایداری و کاهش سنتز هموگلوبین E بهصورت تالاسمی ماژور بتا جلوه میکند.
توضیحات تکمیلی در پیشآمد تولد فرزندی با هاپلوتایپهای تالاسمی دلتا بتا
احتمال بروز کمخونی در فرزندان در نامطلوبترین شرایط در ازدواج ناقل تالاسمی دلتا- بتا با ژنهای دیگر تالاسمی و هموگلوبینوپاتیها مشاهده میشود. تالاسمی δβ یا تالاسمی F اشاره به حذف ژنهای دلتا و بتا روی هاپلوتایپ کروموزوم 11 دارد. در این حالت سنتز جبرانی Hb F ممکن است کــــــــــــمخونیهای هموزیگوت![]() را اندکی آرام و شبیه تالاسمی اینترمدیا درآورد. توجه داشته باشید که در هموزیگوتهای δβ و HPFH حذفی 100% هموگلوبین از نوعF میباشد. آنمی در وراثت همزمان هموگلوبین اچ با ژن تالاسمی ماینور بتا بهصورت اینترمدیا تا ماژور بروز میکند. با توجه به کاهش زنجیره بتا احتمال شکلگیری رسوب β4 در گلبول کمتر است.
را اندکی آرام و شبیه تالاسمی اینترمدیا درآورد. توجه داشته باشید که در هموزیگوتهای δβ و HPFH حذفی 100% هموگلوبین از نوعF میباشد. آنمی در وراثت همزمان هموگلوبین اچ با ژن تالاسمی ماینور بتا بهصورت اینترمدیا تا ماژور بروز میکند. با توجه به کاهش زنجیره بتا احتمال شکلگیری رسوب β4 در گلبول کمتر است.
هموگلوبین لپور اغلب در جمعیت مدیترانه شایع بوده و نتیجه ادغام دو ژن دلتا و بتاست (اسیدآمینههای پایانه آمینی زنجیره دلتا در اسیدآمینههای پایانه کربوکسیلی زنجیره بتا در هم ادغام میشوند). با توجه به اینکه هموگلوبین لپور در ناحیه باند S روی استات سلولز حرکت میکند برای شناسایی آن دو روش الکتروفورز به کار میرود. مقدار هموگلوبین لپور در هموزیگوت لپور از 20% متجاوز نمیشود و با توجه به اینکه پروموترهای ضعیف ژن دلتا در جلوی ادغام δβ قرار میگیرند، سرعت سنتز آن پایین است و در سندرمهای تالاسمی جای میگیرد.
احتمال بروز کمخونی در همراهی وراثت هموگلوبین S با ژنهای تالاسمی و هموگلوبینوپاتیهای دیگر
آزمایش اسکرین ساده، تست حلالیت برای حضور هموگلوبین S است که در بافر فسفاته 2/3 مولار ساپونیندار در حضور سدیم دایتیونات انجام میشود و برای مثبت شدن تست احتیاج به حداقل 20 تا 30 درصد هموگلوبین S است و نتیجه مثبت نیاز به تأئید با الکتروفورز دارد.
ژن داسی در همراهی با ژنهایی که موجب افزایش هموگلوبین F میشوند از قبیل تالاسمی δβ و HPFH بیماری داسی را آرام و حتی بدون علامت میکنند.
همراهی هموگلوبین D پنجاب با ژن داسی آن را تهاجمی و D ایران آرامکننده بیماری است.
وراثت تالاسمیهای آلفا همراه ژن داسی موجب آرام شدن بیماری به علت کاهش سنتز هموگلوبین S میگردد. هتروزیگوت داسی فاقد علائم هماتولوژی و بالینی است.
تهاجمی بودن ژن داسی در رابطه با انواع هاپلوتایپهای ژن داسی متفاوت است. با روش RFLP میتوان نوع تهاجمی ژن داسی را از غیرتهاجمی افتراق داد.
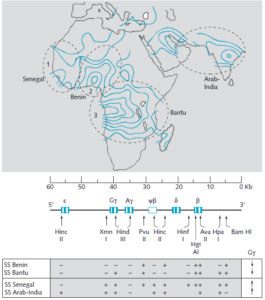
گروهی از آنزیمهای اندونوکلئاز برشدهندهی DNA یا باید خوشهی ژنهای بتا را در جمعیتهای مختلف دنیا در جایگاههای یکسان برش دهند یا برشی ایجاد نکنند، ولی مشاهده میشود که جایگاههای اثرگذاری اندونوکلئازها در مناطق مختلف جهان متفاوت است و بر این اساس، هاپلوتایپهای مختلف، ژن بتا را که حامل جهش داسی است، تعریف میکند. علامت + به معنای جایگاه برش و علامت – به معنای جایگاه بدون برش است.
وراثت همزمان هاپلوتایپ هموگلوبین E با ژنهای تالاسمی و هموگلوبینوپاتیهای دیگر
هموگلوبین E هم ناپایدار است و هم کاهش سنتز دارد و در همراهی با هاپلوتایپ تالاسمی ماینور °β شبیه تالاسمی ماژور درمیآید.
گفتنی است که میزان هموگلوبینهای S ,E و C در حالت هتروزیگوت بستگی به تعداد ژنهای آلفا دارد و برای مثال در هتروزیگوت داسی، فقدان یک و دو و سه ژن آلفا مقدار Hb S به ترتیب 35%، 28% و 21% میشود.
نکته دیگر اینکه هموگلوبین G از واریانت های آلفاست و در اکثر موارد αG همراه با حذف آلفای کناری است و ازاینرو شرایط بالینی بیمار بستگی به جهش G و تعداد حذفها دارد؛ برای مثال ژنوتایپ –/αG – بهصورت هموگلوبین اچ با باند H و S روی استات سلولز درمیآید و –αG/-αG بهصورت تالاسمی ماینورآلفا با باند S روی استات سلولز مشاهده میشود.
واریانت αG همراه با هاپلوتایپ α نرمال بهصورت ααG یا همراه با تالاسمی α+ (-αG) است. در حضور سه ژن سالم آلفا و یک (αα/αGα)αG غلظت HbG حدود 22-20% است. هموگلوبین G تا 30% همراه با ژنوتایپαα/-αG و تا 41 درصد همراه با ژنوتایپα/-αG – است. ژنوتایپ –/ αG – شبیه هموگلوبین اچ است که ایجاد باندهای G و H میکند. توجه داشته باشید که باند G روی باند S استات سلولز قرار میگیرد؛ بنابراین همراهی هموگلوبین G با سایر هموگلوبینوپاتیها بستگی به ژنوتایپ آن دارد.
حدود 80% موارد تالاسمی آلفا ناشی از حذف ژن α میباشد و در 20% موارد علت آن جهشهای ژنی (αT) است که روی هم رفته تهاجمیتر هستند، مانند αCS و αQ که در حالت ααCS/ααCS با وجود اینکه دو آلفای سالم وجود دارد، بیمار علائمدار میشود. در αQ زنجیره آلفا بسیار ناپایدار است.
نکته: در واریانهای زنجیره آلفا چنانچه یک ژن آلفا درگیر شود تنها حدود 20% هموگلوبین غیرطبیعی میشود، مانند αG که در ژنوتایپ ααG/αα تنها 20% هموگلوبین G وجود دارد. از واریانهای دیگر آلفا میتوان به هموگلوبینهای I و آریا اشاره کرد. در هموگلوبین آریا جهش در کد اسیدآمینه شماره47 Asp>Asn رخ میدهد.
هموگلوبین آریا روی استات سلولز روی باند S قرار میگیرد و بایستی از لپور تشخیص افتراقی داد. هموگلوبین آریا غیرتهاجمی است.
واریانهای آلفا ایجاد باند A2 دوبخشی (Split band) میکنند؛ برای مثال هموگلوبین آریا در ناحیه A2 ایجاد دو باند جداگانه میکند و یا هموگلوبین G ایجاد دو باند A2 و G2 میکند. مجموع باندهای A2 یا A2+G2 برابر کل Hb A2 است.
حذف ژنهای خوشه بتا
- حذف تمام یا قسمتی از ژن بتا منجر به تالاسمی β° میگردد، برای مثال یک حذف 619 bp (جفت باز) در انتهای ‘3 ژن بتا شایعترین آلل در جمعیت آسیایی هندی است. حذفهای بزرگ که ناحیه پروموتر ژن بتا را دربرمیگیرد با افزایش غیرمعمول Hb A2 حتی در سطح 9-6 درصد در حالت هتروزیگوت مشاهده میگردد.
- تالاسمی °(δβ) با افزایش 5 تا 25 درصدی هموگلوبین F در هتروزیگوت و 100 درصدی هموگلوبین F در حالت هموزیگوت همراه است.
- تالاسمیهای حذفی (γδβ) و (εγδβ) با کمخونی شدید میکروسیت و هایپوکروم وابسته به تزریق خون در نوزادی همراه بوده و پس از 3 تا 6 ماه شبیه تالاسمی ماینور بتا در بزرگسالان و فاقد علائم بالینی میگردد.
- سندرم HPFH حذفی یا سندرم پابرجایی سنتز هموگلوبین F که در حالت هتروزیگوت میزان هموگلوبین F بین 3 تا 35% متغیر است.
- حذف قسمتی از ژنهای دلتا و بتا و ادغام آنها در نتیجه تداخل نامتقاطع که با سرعت کند ساخته شده و هموگلوبین لپور (α2(δβ)2) نام دارد و در گروه سندرمهای تالاسمی قرار میگیرد.
با توجه به جایگاه ادغام چهار نوع هموگلوبین لپور بنامهای هلندیا، بوستون/واشنگتن، بالتیمور و لیدن شناخته شده است. هموگلوبین لپور در حالت هتروزیگوت 5 تا 15% کل هموگلوبین بوده و در جایگاه S روی استات سلولز قرار میگیرد. با روش الکتروفورز IFE قابل افتراق بوده و در جایگاه ویژه بین A و S قرار میگیرد.
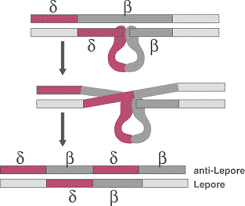
در این شکل ژن هموگلوبین لپور ناشی از کراسینگاور نابرابر ژنهای β و δ طی میوز دیده میشود
- حذف ژنتیکی و ادغام قسمتی از ژنهای γβ، هموگلوبین کنیا را تشکیل داده که مانند HPFH عمل میکند.
برخی از جهشهای غیرحذفی تالاسمیهای آلفا و بتا در جمعیت ایران
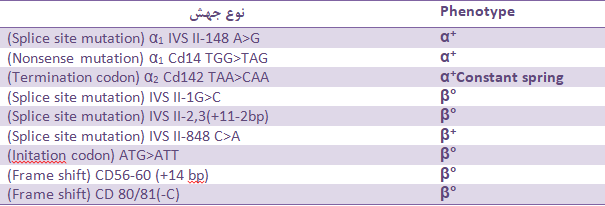
جهشهای گوناگون نقطهای اکثریت موارد تالاسمی °β و +β و حذف ژنی بیشترین علت سندرمهای تالاسمی آلفاست. جهشهای مختلفی عامل ایجاد تالاسمی °β و یا +β است که در جدول زیر به برخی از آنها اشاره شده است.
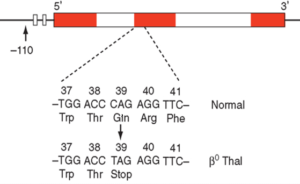
جهش در کد 39 ژن زنجیرهی بتا موجب تالاسمی β0 میشود. کد طبیعی 39 رمز CAG است که اسیدآمینهی گلوتامین (Gln) را رمزدهی میکند. تبدیل کد CAG به TAG رمز خاتمه ایجاد میکند. این جهشهای نقطهای را میتوان با استفاده از پروبهای نشاندار الیگونوکلئوتیدی که مکمل قسمت جهشیافته است یا با روش آرمز (ARMS-PCR) بسهولت تشخیص داد
هتروزیگوت داسی و تالاسمی آلفا
مقدار هموگلوبین S در هتروزیگوت داسی وابسته به تعداد ژنهای آلفاست. در حضور 4 ژن آلفا مقدار هموگلوبین S بین 36 تا 40%، در حضور 3 ژن آلفا بین 34-30% و در حضور دو ژن آلفا بین 28-24% است. چنانچه در حالت هتروزیگوت مقدار Hb S فراتر از 45% شود احتمال ژنوتایپ ααα/αα در همراهی با هتروزیگوت داسی مطرح میشود. در ترکیب داسی و تالاسمی (+Sβ°, Sβ) همیشه هموگلوبین S بیشتر از A است در حالیکه در هتروزیگوت داسی همیشه هموگلوبین A بیشتر از S است. افزایش هموگلوبین A2 ترکیب داسی و تالاسمی بتا و ترکیب داسی و تالاسمی آلفا را همراهی میکند.
مقدار Hb E هم مانند S تحت اثر تعداد ژنهای آلفا قرار دارد. میزان هموگلوبین E بین 30-25 درصد، 25-21 درصد و 21-19 درصد به ترتیب در حضور 4، 3 و 2 ژن آلفا دیده میشود. گاهی همراهی تالاسمی آلفا با هتروزیگوت (AE)E یا هموزیگوتE(EE) و یا β° E/ علامتدار میشود که تحت عناوین بیماری AE Bart و Hb EF Bart از آن یاد میشود. بیماری AE bart disease بارت نتیجه وراثت همزمانی ژنوتایپ اچ (a-/–) با هتروزیگوت E است که با Hb A، Hb E(15-13%) و هموگلوبین بارت همراه است.
شدت این بیماری شبیه هموگلوبین اچ است. همراهی (αCSα/–) با هتروزیگوت E(AE) با کمخونی شدید بالینی همراه است.
از ترکیب ژنوتایپ اچ (a-/–) با هموزیگوت E(EE)هموگلوبینهای E(80%) F(10%), .و Bart(10%) تولید شده و شبیه تالاسمی اینترمدیا شدید میگردد. توجه داشته باشید که ژنوتایپ اچ (a-/–) میباشد و به فرزندان هاپلوتایپهای °α یا +α به ارث میرسد که ممکن است خطری دربر نداشته باشد ولی چنانچه فرزندی علاوه بر ژنوتایپ اچ (a-/–) برای مثال دارای ژنوتایپ °E/β نیز باشد، مبتلا به هموگلوبین EF bart disease و در همراهی با هموزیگوت E(EE) مبتلا به تالاسمی اینترمدیای شدید میگردد. گفتنی است که رسوب زنجیرههای β4S و β4E ایجاد هموگلوبین اچ شبیه توپ گلف نمیکنند.
همراهی ژنوتایپ اچ با تالاسمی هتروزیگوت °β ایجاد کمخونی شدید میکند و در این حالت ممکن است رسوب هموگلوبین اچ به علت کاهش سنتز زنجیره بتا با رنگهای حیاتی مشاهده نشود.
توجه داشته باشید که شخصی ممکن است هاپلوتایپهای سه آلفایی مانند ααα/αα داشته باشد. همراه شدن هاپلوتایپ سه آلفایی برای مثال از پدر و دو آلفایی از مادر در همراهی با ژنوتایپهای β°β یا β+β ایجاد تالاسمی اینترمدیای خفیف میکند.
توجه داشته باشید که ژنوتایپααα/– ممکن است با ناقل خاموش تالاسمی آلفا اشتباه شــــود که در ازدواج با aa/– (تالاسمی هتروزیگوت) ممکن است تولد فرزندی مبتلا به هیدروپس با هموگلوبین بارت را بدنبال داشته باشد.
وراثت ژنهای °α با هموزیگوت HPFH با هموگلوبینهای F و بارت همراهی دارد.
جهش در ژن آلفا را باαT نمایش میدهند؛ برای مثال ژنوتایپ (αTα/αTα) با بیماری هموگلوبین اچ همراه است در حالی که آلفای حذفی مانند (α/-α-) با علائم بالینی همراه نمیباشد، از این رو حذفهای α خطرناکترند. حذفهای آلفا را میتوان با روشهای Gap-PCR و MLPA تعیین کرد.
توجــــه داشته باشیدکه جهش بتا ممکن است بهصورت °β بدون فراورده و یا دارای جهش خفیف (+Mild β) باشدکه 30% ژن سالم فراورده دارد و یا دارای جهش شدید β+(Severe β+) باشد که در این حالت اندکی هموگلوبین A ساخته میشود و همراهی آن با ژنهای داسی یا هموگلوبینهای C و E ایجاد فنوتیپ شدید میکند، در حالی که همراهی جهش خفیف +β با هموگلوبینوپاتیهای دیگر بیماری را خفیف و آرام میکند.
گفتنی است که برخی از جهشهای ژن بتا بهویژه در نیمه دوم اگزون 3 با سنتز زنجیره بتای فوقالعاده ناپایدار همراه است که گاهی ممکن است اثری از زنجیره در سلول یافت نشود و گاهی با زنجیره آلفا ایجاد دایمر کرده که بهسرعت رسوب و تولید انکلوزیون (Inclusion body) در پیشسازهای اریتروئیدی میکند. وراثت هتروزیگوت این نوع ژن بتا که به آن تالاسمی غالب (Dominant) گفته میشود با بزرگی طحال، ژاندیس و آنمی مزمن همراه است. شکل هموزیگوت آن شدیدتر از ° β°β میباشد. افزایش هموگلوبینهای F و A2 از ویژگیهای این نوع تالاسمی است که در شکل هتروزیگوت علائمدار میشود.
در جدول فوق ستون هموگلوبین اچ مربوط به وراثت هموگلوبین اچ با ژنوتایپهای حــذفی آلفا بهصورت (a/aa-) یا بهصورت غیرحذفی از قبیل (ααT/ααT)و یا (ααCS/ααCS) میباشد. از ازدواج این شخص با سندرمهای تالاسمی آلفا و بتا و واریانهای دیگر هموگلوبین، در اکثر موارد خطر کمخونی فرزندان را تهدید میکند.
هموگلوبین H در نوع حذفی دارای هموگلوبین 10-8 gr% با کاهش اندکسهاست و بندرت احتیاج به خون دارد در حالی که نوع حذفی H وابسته به تزریق خون است و گاهی ممکن است حتی موجب تولد فرزندی با هموگلوبینهای H، bart و هیدروپس فتالیس گردد.
فنوتیپهای تالاسمی α° که در جنوب شرق آسیا شایع است با بیماریهای هموگلوبین اچ و بارت همراهی دارد.
حاملگی با جنین هیدروپس در خطر عوارض سقط خودبهخود تولد فرزند مرده ورمکرده ممکن است حاصل ازدواج حاملینα° تالاسمی باشد. در هیدروپس فتالیس زنجیرههای α وجود نداشته و نوزاد در بدو تولد 80% هموگلوبین بارت و 20% هموگلوبین پورتلند 1 و 2 دارد که برای زندگی خارج از رحمی مناسب نیست.
وراثت همزمان ژنوتایپ Hb H با تالاسمی ماینور بتا (β°β) با امکان کمخونی شبیه تالاسمی اینترمدیا با تغییرات شکل و اندازه شدید است که حتی Hb H به علت کاهش زنجیره β ممکن است دیده نشود. وراثت همزمان ژنوتایپ Hb H با هتروزیگوت داسی βSβA با تولید هموگلوبینهای H ,S و A همراه است و شدت بیماری شبیه بیماری هموگلوبین اچ در حالت عادی است. وراثت همزمان ژنوتایپ هموگلوبین اچ با هتروزیگوت E و یا هموزیگوت E به ترتیب با هموگلوبینهای Bart A E و FE bart همراه است که به ترتیب با کمخونی شبیه تالاسمی اینترمدیا و اینترمدیای شدید همراه است. گفتنی است که β4E و β4S ایجاد تترامر H نمیکنند.
همراهی ژنوتایپ اچ با هتروزیگوت HPFH شبیه بیماری H و با هموزیگوت آن ایجاد هموگلوبینهای F و bart میکند.
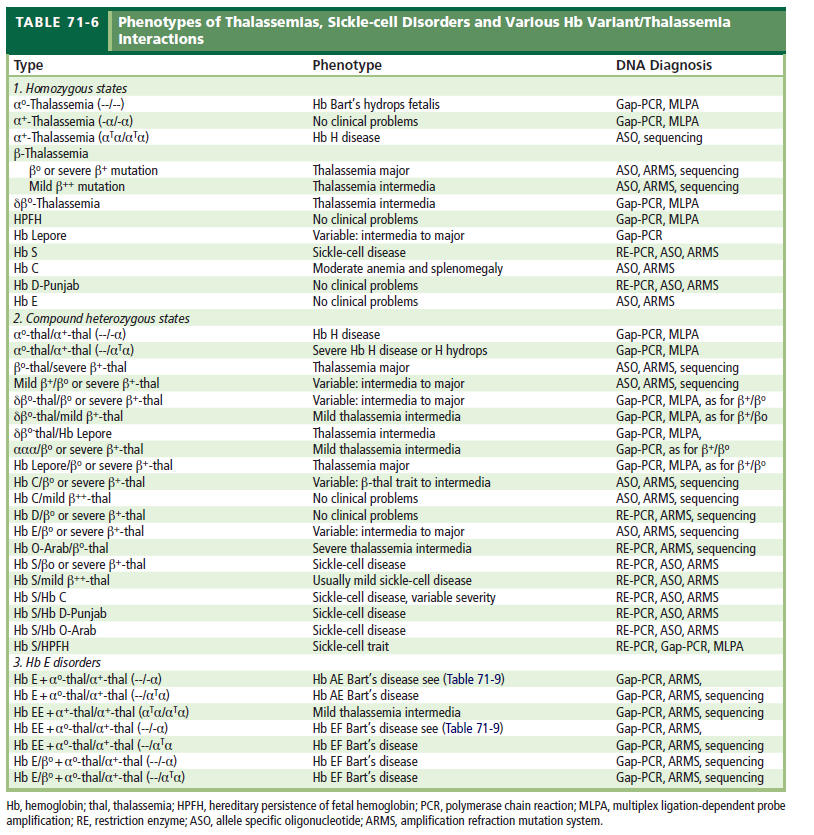
طبقهبندی بالینی و ژنتیکی تالاسمیها و تشخیص آنها
هموگلوبینهای M
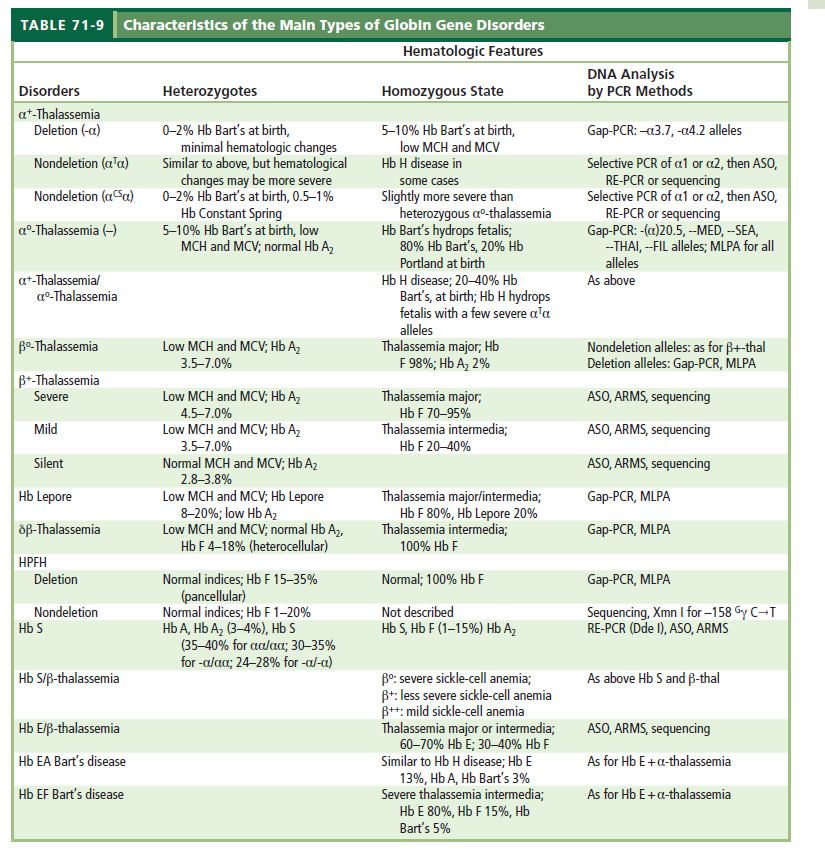
چنانچه یکی از والدین مبتلا به متهموگلوبینمی (Hb M) باشد به شیوه اتوزوم غالب به فرزندان منتقل میشود. تاکنون 7 نوع Hb M شناخته شده است که جهش در زنجیرههای α یا β و γ در اکثر آنها موجب جایگزینی تیروزین بجای هیستیدین میشود و حلقه هیم در حالت فریک (+Fe3) پایدار شده و قادر به حمل اکسیژن نمیباشد. جهشهای زنجیرههای گاما و آلفا با سیانوز در بدو تولد و جهش بتا با سیانوز در 6 ماهگی مشخص میشود. کاهش آنزیم Diaphorase 1 بهصورت اتوزوم مغلوب نیز موجب متهموگلوبینمی میگردد. هموگلوبین M در اکثر موارد ایجاد باند خاصی در الکتروفورز نکرده و تشخیص سیانوز ناشی از Hb M برای افتراق سیانوز ناشی از بیماریهای قلبی- ریوی مهم است. جذب ماکزیمم Hb M در طولموج 630 نانومتر برای تشخیص کمککننده است.
هموگلوبینهای ناپایدار
ابتلای پدر یا مادر به هموگلوبین ناپایدار موجب انتقال آن به فرزندان میشود. تاکنون 300 نوع هموگلوبین ناپایدار شناخته شده است. جهش موجب دناتوره شدن و بهم خوردن ساختمان آلفا هلیکس زنجیره گلوبین میگردد. برخی جهشها موجب جدا شدن دایمرهای گلوبین و جدا شدن ساختار هیم و یا تولید پاکت هیدروفیل در اطراف حلقه هیم میگردد. جهشهای ناپایدار بتا بیشتر از آلفا علائمدار میگردد. گلوبینهای دناتوره شده ایجاد هاینزبادی و کمخونی همولیتیک با ژاندیس و بزرگی طحال و گاهی سیانوز میکنند. دفع ادراری قهوهای رنگ حاکی از مشتقات دایپرول در بیماران است. همراهی ژن بتای ناپایدار با تالاسمی °β و +β ایجاد کمخونی شدید میکند. هموگلوبینهای ناپایدار در حالت هتروزیگوت هم علائمدار هستند. برخی از واریانتهای ناپایدار β چنان ناپایدار هستند که بهسرعت از بین رفته و تنها مطالعه DNA قادر به تشخیص آنها میباشد. آزمایشهای رسوب هموگلوبین در ایزوپروپانول 17% و تست حرارتی از آزمایشهای اسکرین تشخیص هموگلوبینهای ناپایدار هستند. هموگلوبینهای ناپایدار دارای میل ترکیبی کم یا زیاد یا نرمال برای اکسیژن هستند.
جهش ناپایدار در یک ژن آلفا با تولید 5 تا 20% هموگلوبین ناپایدار همراه است که علائم بالینی ندارد ولی جهش ناپایدار بتا با تولید 20 تا 40 درصد هموگلوبین ناپایدار علائمدار میشود.
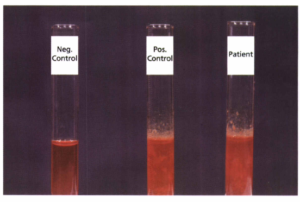
آزمایش ایزوپروپانول 17%؛ در این آزمایش به 2 سیسی محلول بافری ایزوپروپانول که از قبل به 37 درجه رسیده است، 2/. سیسی همولیز اضافه میشود و در 37 درجه گذاشته میشود و در فواصل 5، 20 و 30 دقیقهای از نظر رسوب بررسی میگردد
هموگلوبینهای با میل ترکیبی کم یا زیاد برای اکسیژن
جهشهایی که موجب بر هم خوردن واکنش حلقه هیم با یکدیگر یا اثر بوهر یا اتصال 2,3DPG به زنجیره بتا یا پایدار شدن هموگلوبین در وضعیت دیاکسی میگردند با میل ترکیبی کم یا زیاد برای اکسیژن همراهی دارند.
واریانتهای با میل ترکیبی زیاد، منحنی اشباع اکسیژن را بهطرف چپ و با میل ترکیبی کم منحنی را بهطرف راست سوق میدهد که نتیجه آن به ترتیب کاهش پسدهی و افزایش پسدهی اکسیژن است.
به علت شیوع کم، اکثر موارد ارثی هتروزیگوت و فاقد علائم است و چنانچه هموگلوبین با میل ترکیبی زیاد علائمدار شود با پلیسیتمی و افزایش شمارش گلبولهای قرمز بروز میکند، در حالیکه هموگلوبینهای با میل ترکیبی کم منجر به آنمی و سیانوز میشود چون سریع رها شدن اکسیژن از هموگلوبین موجب کاهش اکسیژن بافتها میگردد.
اندازهگیری P50 (فشاری از اکسیژن که 50% اکسیژن اشباع است) از راههای تشخیص و افتراق است. میزان نرمال P50 بین 35-17 میلیمتر جیوه و کمتر از 17 بیانگر میل ترکیبی زیاد و بیشتر از 35 بیانگر میل ترکیبی کم است.
مزایا و معایب روشهای مختلف الکتروفورز هموگلوبین
الکتروفورز روی استات سلولز در PH قلیایی (CAE)
مولکولهای هموگلوبین در PH قلیایی دارای شارژ منفی متفاوت شده و بهسوی آند با سرعتهای مختلف روی غشای استات سلولز حرکت میکنند. به وسیله مقایسه با حرکت استانداردهای هموگلوبین میتوان باندهای ایجادشده را بهطور اولیه افتراق داد. این روش الکتروفورز قادر به جداسازی هموگلوبیــــــــــــنهای Hb A2/C/E/O ,Hb S/G/D/Lepore , F ,A و هموگلوبینهای گروه فاست (با حرکت سریع) J/bart/H میباشد. برای جداسازی باندهایی که در یک گروه قرار میگیرند نیاز به آزمایشهای تکمیلی یا استفاده از روش الکتروفورز مکمل است؛ برای مثال آزمایش حلالیت قادر به افتراق هموگلوبینS از بقیه گروه همشارژ است و یا الکتروفورز روی ژل آگاروز در PH اسیدی میتواند هموگلوبینهای C وS را از بقیه جدا کند. اندازهگیری دانسیتهمتری A2 دقیق نبوده و نیاز به کروماتوگرافی ستونی یا روشهای دیگر دارد.
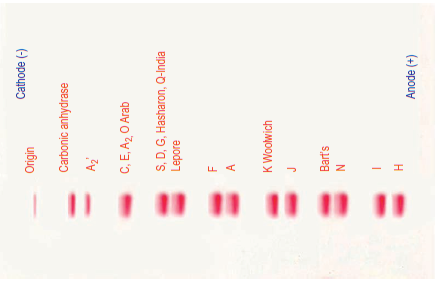
جایگاه قرارگیری هموگلوبینهای مختلف بر روی استات سلولز در PH قلیایی
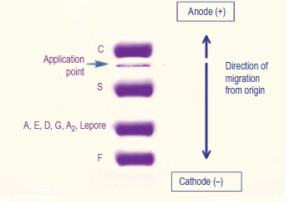
جایگاه قرارگیری هموگلوبینهای مختلف بر روی ژل آگارز در PH اسیدی
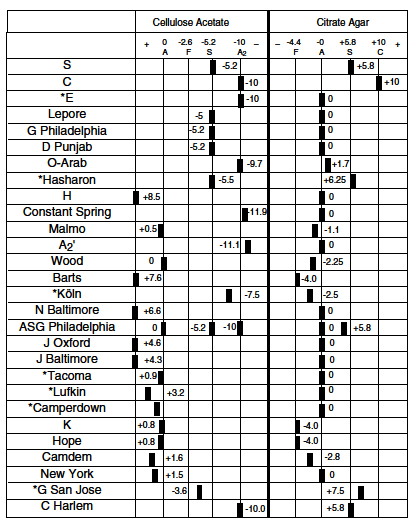
در جدول، الگوهای الکتروفورز هموگلوبین بر روی استات سلولز و ژل آگارز مشاهده میشود. جایگاه هموگلوبین A بهمثابهی شاخص اصلی است و از این رو آن دسته از هموگلوبینها که حرکتی کندتر از A دارند در گروه حرکت کُند و آن دسته که جلوتر از A قرار میگیرند در گروه هموگلوبینهای سریع طبقهبندی میشوند. چنانچه شاخص یا جایگاه هموگلوبین A عدد صفر فرض شود، فاصلهی هموگلوبینها از شاخص با علامتهای + و – مشخص میشود؛ مثلاً هموگلوبین H با عدد 8/5+ سریعتر از هموگلوبین بارت با 7/6+ است.

آزمایش حلالیت برای دستیابی به نتیجهی مثبت به حداقل 20 درصد هموگلوبین S نیاز دارد. افزودن گلبولهای قرمز حاوی هموگلوبین S به بافر فسفاته ساپونیندار با مولاریتهی 3/2 موجب کدر شدن و مانع از مشاهدهی خطوط کارت در پشت لوله میشود.
الکتروفورز با استفاده از نقطه ایزوالکتریک (Iso Electric Focusing) IEF
در این روش مولکولهای هموگلوبین توسط جریان الکتریسیته در یک شیب PH با توجه به نقطه ایزوالکتریک با افتراق شفاف جدا میشوند. هر مولکول پروتئینی در PH مخصوص مجموع بارهای مثبت و منفی آن برابر و شارژ آن صفر شده و از حرکت متوقف میشود که به آن نقطه PI (ایزوالکتریک)گفته میشود. روشهای حساس IEF قادر به جداسازی پروتئینها حتی با تفاوت PI در 0/01 واحد PH میباشند.
مزایای IEF
- جداسازی واریانتهای مختلف هموگلوبین
- ایجاد باندهای شفاف
- قابل استفاده برای قطرات خون خشکشده
- جداسازی خوب هموگلوبینهای O ,E ,D ,C ,S ,F ,A
- افتراق هموگلوبین بارت
معایب IEF
- جداسازی مشتقات پس از ترجمه هموگلوبین
- غیرقابل افتراق بودن Hb A2/E /D-punjab
- اندازهگیری A2 قابل اعتماد نیست
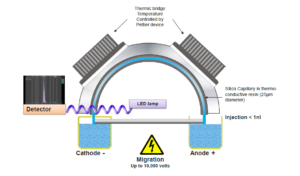
الکتروفورز کاپیلاری (CE)
الکتروفورزکاپیلاری دارای قابلیت جداسازی بسیار خوب انواع هموگلوبین در شرایط قلیایی با استفاده از بافر قلیایی در لولههای موئینه شیشهای از جنس سیلیکا میباشد. تحت اثر یک ولتاژ قوی حــدود 9800 V، مولکولهای مختلف هموگلوبین با توجه به شارژ با سرعتهای گوناگون بهطرف آشکارساز حرکت میکنـــــند. ولتاژ بالا موجب جداسازی شفافتر هموگلوبین در مقایسه با استفاده از ولتاژ 350-310 در روشهای عادی است. مولکولهای هموگلوبین نسبت به باندهای A1 و A2 با استفاده از جذب فتومتری در طولموج 415 نانومتر استاندارد میگردند و امکان اندازهگیری مســــــــــتقیم Hb F ,Hb A و Hb A2 را فراهم کرده و قادر به جداسازی هموگلوبینهای D ,C ,S پنجاب،G فیلادلفیا و هموگلوبین E بوده و هموگلوبین لپور و E را از Hb A2 افتراق میدهد. در مقایسه با HPLC افتراق هموگلوبین اچ سختتر است و روی هم رفته نیاز به روش دیگر و مکمل برای تأئید جداسازی هموگلوبین میباشد.
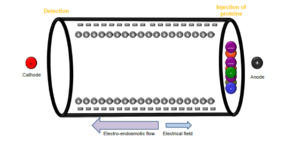
الکتروفورز کاپیلاری
تحت اثر ولتاژ قوی مولکولهای مختلف هموگلوبین در بافر جداسازی از هم جدا میشوند. مولکولها در جهات مختلف حرکت کرده و توسط آشکارساز که معمولاً در انتهای کاپیلاری است شناسایی میشوند
مزیت روش CE
- نیمه اتوماتیک
- جداسازی بسیار خوب
- نتایج فوری
- نیاز به نمونه کم
- اندازهگیری A2 ,F ,A
- افتراق Hb A2 /E/S
- افتراق اولیه Hb S-C-D-G-E
- جداسازی HbLepore /E /C /A2
- جداسازی هموگلوبین کنستانت اسپرینگ (جهش در کد پایانی ژن α2 که منجر به سنتز زنجیره آلفا طویل شده با 31 اسیدآمینه اضافه میگردد). این متد روش انتخابی برای تشخیص Hb CS میباشد.
معایب CE
- دشواری تشخیص Hb H
- نیاز به افزودن مواد کنترل چنانچه Hb A موجود نباشد.
کروماتوگرافی مایع با قابلیت بالا (HPLC)
HPLC متد انتخابی برای جداسازی ایدهآل، تشخیص صحیح و اندازهگیری کمی هموگلوبینها است. تمام انواع هموگلوبینها روی ماتریکس جامد تعویض یونی پیوند خورده و سپس در رابطه با زمان و تغییر PH و افزایش قدرت یونی از قبل طراحیشده، هموگلوبینهای مختلف در زمان مشخص (Retension Time) از ستون جدا و با اندازهگیری جذب نوری در مقایسه با استاندارد اندازهگیری میشوند. اندازهگـــــــــیری Hb S ,Hb A2 و Hb F بسیار دقیق بوده و برای شناسایی ناقلین تالاسمی بتا مناسب است، گرچه میتوان واریانهای مختلف هموگلوبین را شناسایی کرد. سیستم HPLC در نبود هموگلوبینهای S و E مقدار A2 را بهطور دقیق اندازهگیری میکند و از این رو مشکل تشخیصی با هتروزیگوت ترکیبی E/B و هموزیگوت EE ایجاد میکند. هموگلوبین لپور نیز با هموگلوبین لپور نیز با Hb A2 جدا شده و تفسیر را مشکل میسازد.
مزایای HPLC
- تمام اتوماتیک
- جداسازی خوب هموگلوبینهای D ,C ,S ,F ,A2 ,A پنجاب و G فیلادلفیا
- اندازهگیری دقیق A2
- هموگلوبینهای بارت و H قابل تشخیص بوده ولی قابل اندازهگیری نیستند.
- تشخیص هموگلوبین کنستانت اسپرینگ
معایب
- نیاز به کالیبراسیون دارد
- قادر به جداسازی E/Lepore/A2 نیست
- نواقص ستون
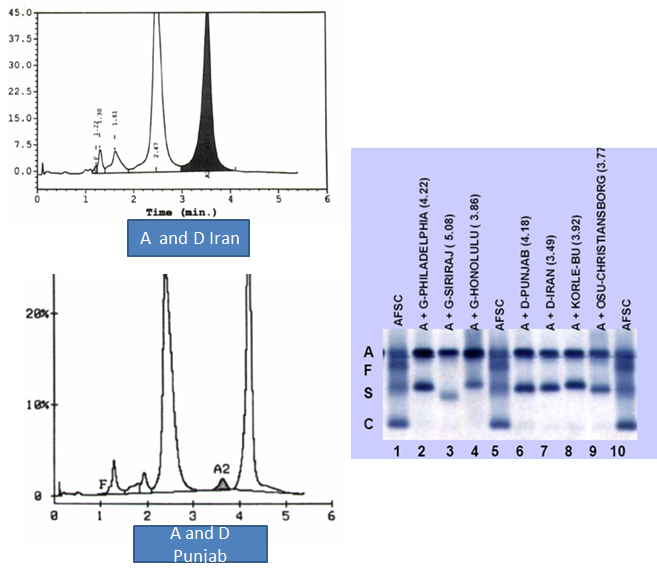
کروماتوگرام هموگلوبینهای D ایران و D پنجاب با HPLC و الکتروفورز آنها بر روی استات سلولز مشاهده میشود. زمان جدا شدن هموگلوبینها از ستون HPLC در پرانتز قید شده است
تشخیص قطعی جهشها
چندین روش بر مبنای PCR برای تشخیص قطعی ناقلین تالاسمی فراهم شده است. این روشها شامل هیبریدی شدن با الیگونوکلئوتیدهای اختصاصی آلل (ASO-hybridization) و آنالیـــــــــــــز
RDB, Gap-PCR, ARMS-PCR و تعیین مستقیم توالی نوکلئوتیدها(Sequencing) میباشد. گفتنی است که با وجود بیش از 200 جهش شناختهشده تالاسمی بتا، تنها تعداد محدودی از جهشها مسئول بیشتر از 90% تالاسمیهای یک منطقه جغرافیایی هستند و از این رو طراحی پرایمر (Primer) و پروب (Probe)که جهشها را هدف قرار دهند، چندان مشکل نمیباشد. آنالیز RDB از روشهای شایع برای تأئید جهشهای تالاسمی بتا میباشد که پروبهای الیگونوکلئوتیدی مکمل آلل جهش یافته و نوع سالم آلل را بر روی غشا قرار داده و با محصول PCR مجاور میگردد. برای جهشهای ناشناخته با استفاده از تعیین توالی با استفاده از آمپلیکنAmplicons) ) انجام میگیرد. برای شناسایی ژنهای حذفشده تالاسمی آلفا میتوان در صورت شناسایی نقطه شکسته از gap-PCR استفاده کرد و چنانچه تالاسمی آلفا بسیار مشکوک باشد ولی gap-PCR جواب قطعی بدست نداده است، استفاده از Real Time PCR ممکن است سودمند باشد. با این روش میتوان حذفهای آلفا که محل شکستگی واضحی ندارند و نیز هاپلوتایپهای سه آلفایی را تشخیص داد. روش(Multiplex ligation probe amplification) MPLA قادر به آشکار کردن حذفهای ناشناخته چند صد جفت بازی میباشد. برای شناسایی جهشهای نقطهای میتوان از آنالیز RDB و ARMS و تعیین سکانس مستقیم استفاده کرد.
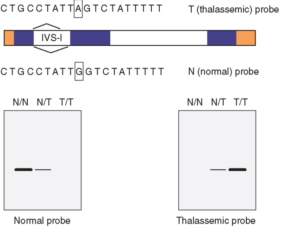
استفاده از پروب های ASO برای تشخیص تالاسمی β. دو پروب الیگونوکلئوتیدی فقط در موقعیت جهش تفاوت دارند
تشخیص ژنتیکی قبل از لانه گزینی تخمک بارور شده (Pre implantation) با IVF ممکن است قادر باشد تا تولد فرزندی سالم از ناقلین را ارائه دهد. روشهای جدید مانند NGS با اسکرین کردن طیف وسیعی از اختلالات ژنتیکی مغلوب (Pan ethnic) از قبیل تالاسمی، چالشهای مشورتی را ممکن است بدنبال داشته باشد.
مروری بر انواع تالاسمیها و هموگلوبینوپاتیها و تشخیص آنها
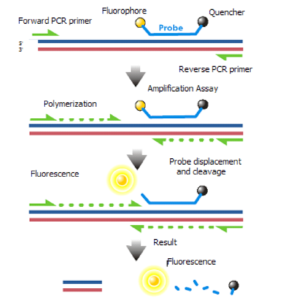
. مکانیسم شیمیایی real time PCR Taq Man
پروبهای Taq Man از یک بخش الیگونوکلئوتیدی، یک ریپورتر یا فلوروفور و یک quencher تشکیل شده است. در حالت طبیعی quencher از فلورسانس فلوروفور ممانعت میکند. متعاقب پلیمریزاسیون DNAی تکرشتهای، فعالیت اگزونوکلئازی Taq پلیمراز سبب شکستن پروب گردیده، فلوروفور آزادشده و فلورسانس آن ساطع میشود. میزان فلورسانس دتکت شده در این روش متناسب با فلوروفور آزاد شده و مقدار الگوی DNA در PCR است
مروری بر روشهای تشخیصی پیش از تولد تالاسمی
متابولیسم پروتئینها و اسیدهای آمینه و اختلالات ناشی از آنها
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام