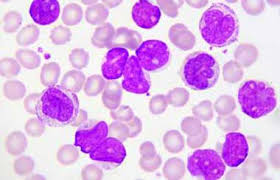
لوسمی حاد لنفوبلاستیک و تازههای آن در ایران
ملیحه نجفپور، دانشجوی کارشناسی ارشد خون شناسی و بانک خون –دانشگاه علوم پزشکی تبریز
Mmahsa_22@yahoo.com
تعریف:
واژه لوسمی حاد به بدخیمیهای هماتولوژیک که با افزایش تعداد بلاستهای رده میلوئید یا لنفوئید همراه است اطلاق ميشود. اصطلاح حاد به شروع سریع و به سرعت کشندهای اشاره میکند که ماهیت تمایز نیافتهای از سلولهای لوسمیک را نشان میدهد.
لوسمی شایعترین سرطان رایج در بین کودکان است و لوسمی حاد لنفوبلاستیک شایعترین زیرگروه میباشد و 80-75% موارد را شامل میشود (1).
لوسمی حاد لنفوبلاستیک (Acute Lymphoblastic Luekemia:ALL) در درجه اول بیماری کودکان محسوب میشود که با افزایش وقوع بین سن 3-2 سال همراه است، سپس شیوع آن کاهش مییابد به طوری که در سن بین 50-25 سالگی نادر میباشد و بعد از آن مجدداً افزایش مییابد تا به دومین اوج شیوع خود (اما با شیوع کمتر) در سن بیشتر از 80 سالگی میرسد (1).
با پیشرفتهای اخیر در درمان این لوسمی میزان بهبودی تا 80% رسیده است.
اپیدمیولوژی
احتمال وقوع ALL در ایران سالانه1 /6 در 100000 مرد و 5/2 در 100000 زن تخمین زده میشود (2). متوسط سن تشخیص 13 سال میباشد و تقریباً 60% موارد در سن زیر 20 سال تشخیص داده میشوند (1). ALL شایعترین بدخیمی در افراد زیر 15 سال است به طوریکه 23% از انواع سرطانها و 76% از همه انواع لوسمیها در این گروه قرار دارند (1). وقوع آن در پسران بیشتر از دختران است و در موارد T-ALL این وقوع 4 برابر بیشتر است. در سال اول زندگی احتمال وقوع در دختران حدود 1/5 برابر نسبت به پسران بیشتر است. در کشورهای صنعتی احتمال وقوع در نژاد اروپایی بیشتر از نژاد آفریقایی میباشد. در کشورهای پیشرفته بالاترین میزان وقوع آن در سن 5-2 سال است. شیوع در سفید پوستان بیشتر از سیاه پوستان است (1). جمعیت کوچکی از بیماران مبتلا به ALL کودکان، بیماریهای ژنتیکی زمینهای دارند. کودکان مبتلا به سندرم داون 30-10 مرتبه افزایش خطر برای ALL دارند. جهش در JAK2 در سلولهای ALL کودکان با سندرم داون دیده میشود. از سایر اختلالات ژنتیکی میتوان به آتاکسی تلانژکتازیا و سندرم بلوم اشاره کرد (1). دوقلوها نسبت به کودکان غیر دوقلو 4-2 برابر خطر بیشتری برای لوسمی در طول دهه اول زندگی خود دارند. در دوقلوهای همسان، وقتی لوسمی در یکی از کودکان بروز میکند احتمال وقوع آن در کودک دیگر 20% است، با این وجود این احتمال میرود که انتقال ALL در دوران جنینی از طریق گردش خون مشترک جفت صورت گرفته باشد (1). وقتی لوسمی در سن زیر یک سال تشخیص داده شود در مدت چند ماه ممكن است در خواهر یا برادر دوقلوی او دیده شود. در دوقلوهای همسان با t(4;11)/MLL-AF4 این دوره کوتاهتر میشود، برخلاف آن در دوقلوهایی با ETV6-RUNX1 یا فنوتیپ سلول T دوره نهفتگی طولانیتر است و نیاز به وقایع ژنتیکی مضاعف برای ایجاد لوسمی دارد. وقوع ALL هیپردیپلوئید قبل از تولد و در دوران جنینی دیده شده است اما نیاز به رویدادهای بعد از تولد برای ترانسفورم شدن به سمت بدخیمی دارد. ALL با t(1;19)/TCF3-PBX1در اکثر موارد منشأ بعد از تولد دارد (1).
طبقه بندی:
دو تقسیم بندی که به طرز گستردهای مورد استفاده قرار میگیرد، طبقه بندی FAB و WHO میباشد، که هر کدام از آنها ساختارهای طبقه بندی خاص خود را دارد. به طور کلی هر دو نوع این طبقه بندی، لوسمیهای حاد را بر اساس منشأ سلولهای بلاست، به دو نوع میلوئید و لنفوئید طبقه بندی میکنند (3).
در طبقه بندی FAB، ALL بر اساس مرفولوژی سلولهای بلاست به سه زیرگروه L1 تا L3 طبقه بندی میشود. زیرگروه L3 در 5% ALL بزرگسالان دیده میشود و تنها زیرگروهی است که با علائم بالینی همراه است زیرا در این زیرگروه سلولهای B بالغ درگیر میشوند و پروتکل درمانی خاص خود را دارد. تشخیص این زیرگروه باید با آنالیز مارکرهای سطحی تأیید شود (3).
طبقه بندی FAB نمیتواند تشخیص درستی بین ALL و لوسمی حاد غیر لنفوئید بدهد و هیچ پیشآگهی و درمان مرتبط با درمانهای معاصر را نمیدهد، با این وجود طبقه بندی لوسمیهای حاد اخیراً بر پایه WHO صورت میگیرد (3).
برای طبقه بندی ALL بر اساسWHO نیاز به آنالیز مرفولوژی، ارزیابی سیتوشیمی، ایمنوفنوتیپ و سیتوژنتیک است (3).
بر طبق طبقه بندی WHO، ALL همــــــراه با لنفوم لنفوبلاستیک- نئوپلاسم در پروکورسرهای لنفوئید- طبقه بندی میشود. این طبقه بندی شامل B lymphoblastic leukemia/lymphoma،
B lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities،
و T lymphoblastic leukemia/lymphoma میشود. در این طبقه بندی mature B-ALL به عنوان زیرگروه جدا محسوب نشده و به عنوان لنفوم بورکیت که به صورت نئوپلاسم B بالغ است محسوب میشود (3).
رنگهای سیتوشیمی مانند پریودیک اسید شیف در بیش از 70% موارد ALL مثبت میشود، در حالی که میلوپراکسیداز، سودان بلاک، استرازهای اختصاصی و غیر اختصاصی به طور معمول منفی میباشند (3).
طبقه بندی ALLT با استفاده از تکنیکهای ایمنولوژیکی (ایمنوفنوتیپ)- استفاده از آنتیژنهای اختصاصی مثل مولکولهای داخل سیتوپلاسمی یا سطح سلول در طی مراحل تمایز- برای تعیین رژیم درمانی برای بیماران مناسب است (3). (جدول 1)
Immunological classification, corresponding cytogenetic and molecular aberrations and frequencies in ALL
| Molecular genetics | Cytogenetic† | Surface marker | |
| HLA – DR + , TdT+ , CD19 + and/or
CD79a + and/or CD22 |
B- lineage | ||
| ALL1 ( MLL ) – AF4 | t(4;11) | No further differentiation markers | Pro B- ALL |
| BCR – ABL | t(9;22) | CD10 + | Common- ALL |
| BCR – ABL , E2A – PBX1 | t(9;22), t(1;19) | CD10 +/ , cyIgM+ | Pre B – ALL |
| MYC – IGH | t(8;14) | CD10 +/ , sIgM+ | B – ALL |
| TdT+ , cyCD3 + or sCD3 + | T – lineage | ||
| LMO1 – TCR α / δ | t(11;14) | cyCD3 + , CD7 + , CD5 +/ , CD2 +/ | Early T – ALL |
| HOX11 – TCR α / δ | t(10;14) | cyCD3 + , CD7 + , CD1a + , sCD3 +/ | Cortical T – ALL (Thy ALL) |
| sCD3 + , CD1a | Mature T – ALL |
cyIgM, cytoplasmic IgM; sIgM, surface IgM; cyCD3, cytoplasmic CD3; sCD3, surface CD3; TdT, terminal deoxynucleotidyltransferase
نقش بررسی سیتوژنتیک در ALL بزرگسالان اهمیت بسیاری دارد و جزء متغیرهای پیشآگهی مستقل در پیشبینی وضیعت لوسمی میباشد. دسته بندی ALL بر اساس ناهنجاری سیتوژنتیک بر طبق زیر میباشد:
ALL هیپردیپلوئید و هیپودیپلوئید
تقریباً در نیمی از موارد سلولهای لوسمیک هیپردیپلوئید هستند و این موارد با شمارش کروموزومی 60-51 کروموزوم و پیشآگهی ضعیف همراهند. معمولاً B-ALL هستند و لنفوبلاستها با این کاریوتیپ گرایش به آپوپتوز و تجمع بیشتر متوتروکسات و متالوبیتهای فعال پلیگلوتامات دارند. در بین مواردی که افزایش تعداد کروموزوم دیده میشود، تنها ترایزومی کروموزوم 4 و 10 و 17 در مطالعات با پیشآگهی خوب همراه بوده است. موارد تریپلوئیدی (81-69 کروموزوم) پاسخ درمانی مشابه با موارد ALL غیر هیپردیپلوئید دارند. مواردی که تتراپلوئید هستند (94-82 کروموزوم) فنوتیپ T-ALL را نشان میدهند. هیپودیپلوئیدی (کروموزوم کمتر از 45) در کمتر از 2% موارد ALL دیده میشود و با پیشآگهی ضعیف همراه است (3).
ALL با بازآرایی TEL-AML1(ETV6-RUNX1) یا t(12;21)(p12;q22)
تقریباً در 20% موارد دیده میشود و بیماران با این فیوژن ژنی از لحاظ بالینی به گروههای مختلفی تقسیم میشوند و به طور معمول این افراد بین سن 7-2 سال بوده و کاریوتیپ دیپلوئید دارند. برخی مطالعات نتایج بالینی با اهمیتی نداشته اما در اکثر مطالعات پیشآگهی بسیار خوبی داشته است. ژن ETV6 به خانواده Ets از فاکتورهای نسخه برداری تعلق دارد و نقش اساسی در هماتوپوئز دارد. RUNX1 فاکتور نسخه برداری را کد میکند که مانند یک هترودایمر به وسیلهی CBF-ᵝ(core binding factor) به DNA وصل میشود و برای پیشرفت هماتوپوئز نهایی ضروری است. اخیراً نشان داده شده است که پروتئین ETV6-RUNX1 به Smad3 وصل میشود-که هدف اصلی سیگنالینگ TGF-ᵝ میباشد- و توانایی آن را در فعال کردن پروموتورهای هدف تغییر میدهد و نتیجه آن کاهش حساسیت اثر مهاری TGF-ᵝ است. تقریباً در 90% موارد ALL با این فیوژن ژنی، الل ETV6 به طور کامل یا نسبی در جمعیت متغیری از سلولهای توموری حذف میشود و این امر میتواند یک رویداد ثانویه مهم برای لوکموژنزیس باشد؛ علاوه بر این، این جابجایی در برخی موارد T-ALL نیز دیده شده و حضور آن در سطح پروژنیتورهای بند ناف منجر به کاندیدا شدن این سلولها به سمت روند پریلوسمیک میشود. فیوژن مزبور با روش FISH یا RT-PCR قابل شناسایی است (3).
ALL با بازآرایی E2A-PBX1(TCF3-PBX1) یا t(1;19)(q23;p13)
تقریباً در 25-20% موارد pre B-ALL دیده میشود. در موارد B-ALL معمولاً رایج نيست و در کمتر از 5% موارد در اطفال و در موارد نادری در بزرگسالان دیده میشود. ژن TCF3(E2A) فاکتورهای نسخه برداريی را کد میکند که در تکامل لنفوسیت B و میوسیت نقش دارند. PBX1 نیز یک فاکتور نسخه برداری متصل شونده به DNA است که به طور نرمال در سطح سلولهای لنفوئید بیان نمیشود. وقتی فیوژن TCF3-PBX1 صورت میگیرد ناحیه اتصال PBX1 به DNA توسط TCF3 اشغال میشود و باعث تمایز پروژنیتورهای B سل به سمت روند نئوپلاستیک میشود. این جابجایی در بیماران کم سن با پیشآگهی ضعیف همراه است، اگرچه افراد به درمان اولیه پاسخ خوبی میدهند اما هنوز افزایش احتمال خطر عود لوسمی در CNS را دارند (3).
یکی دیگر از فیوژنهای ژنی E2A، t(17;19)(q22;p13) است. در این جابـــجایی E2A با ژنی که فاکتور نسخه برداری HLF (Hepatic Leukemia Factor) را کد میکند ادغام میشود. بیماران با این جابجایی با افزایش میزان Ca و کواگولوپاتی و پیشآگهی ضعیفی همراه هستند (3).
ALL با بازآرایی ژن MLL
MLL یک پروتئین متصل شونده به DNA است که دارای چندین دومین فعال از نظر عملکردی میباشد و نقش یک فاکتور نسخه برداری چندگانه با فعالیت هیستون متیل ترانسفرازی را داراست. ژن MLL پروتئین متصل شونده به DNA را کد میکند که بیان بسیاری از ژنها از جمله ژنهای خانواده HOX را تنظیم میکند؛ که این ژنها در حفظ و پیشبرد پروژنیتورهای میلوئید و لنفوئید نقش دارند. MLL برای روند هماتوپوئز و پیشبرد دوران جنینی حیاتی بوده و پروتئین فیوژن شده MLL میتواند منجر به ترانسفورم شدن سلولهای هماتوپویتیک به سمت رده لوسمیک شود.t(4;11)(q21;q13) شایعترین ناهنجاری ژن MLL است و باعث ادغام ژنی MLL با AFF1 میشود و معمولاً در لوسمی نوزادان و بیماران با زیرگروه pro B-ALL دیده شده و احتمال وقوع آن در بزرگسالان 5% است. بیماران از لحاظ بالینی با شمارش لکوسیت بالا، ارگانومگالی و درگیری CNS همراهند. نتایج درمان با توجه به سن متفاوت بوده به طوری که در نوزادان با بدترین پیشآگهی همراه است. بیان برخی ژنها منجمله FLT3، LMO2، HOX در موارد ALL با بازآرایی MLL افزایش مییابد که در حالت نرمال در سطح ردهی غیر لنفوئید بیان میشوند. برای تشخیص این ناهنجاری از روش RT-PCR و ساترن بلات و FISH استفاده ميشود (3).
ALL با بازآرایی BCR-ABL1 (کروموزوم فیلادلفیا مثبت (PH+)
این فیوژن ژنی منجر به تولید ABL تیروزین کیناز ناقص و ترانسفورم شدن سلولهای هماتوپویتیک به سمت بدخیمی میشود. در 30-20% بزرگسالان و تنها در 3% کودکان دیده میشود. دو ناحیه شکست متداول در فیوژن ژنی BCR-ABL دیده میشود، که در اکثـــر موارد B-ALL کودکان (90-80%) شکست در ناحیه minor-BCR(190KD protein) وجود دارد که منجر به تولید e1-a2 BCR-ABL Mrnaمیشود. تقریباً يك سوم موارد بزرگسالان نسخههای e13-a2 یا e14-a2 را نشان میدهند که مربوط به شکست در ناحیه (210 KD protein) Major-BCR میباشد و دو سوم شکست در minor-BCR را نشان میدهند. ALL با BCR-ABL مثبت با پیشآگهی ضعیف در شیمی درمانی همراه است. پیشرفت در مهار کنندههای تیروزین کیناز منجر به ناتوانی مسیرهای مولکولی رشد سلولهای لوسمیک میشود. در PH+ALL بزرگسالان، ایماتینیب در ترکیب با شیمی درمانی معمولی سطح بهبودی تا 95% میدهد، البته در اغلب موارد حفظ این پاسخ مشکل است. اخیراً گزارشی نشان داده که شیمی درمانی ترکیبی با ایماتینیب میزان EFS (event free survival) سه ساله را تا 80% در کودکان بالا برده است. اکثر موارد ALL با این فیوژن ژنی نیاز به درمان تهاجمیتر شامل پیوند استم سل آلوژنیک دارد. روش شناسایی این ناهنجاری ژنتیکی RT-PCR میباشد (3).
علاوه بر B-ALL برخی ژنها که در T-ALL دچار بینظمی می شوند شامل SCL(TAL-1)، LMO1(TTG-1)، LMO2(TTG-2) و HOX11 میباشند. از سایر موارد حذف کروموزوم 9P21 ناحیهی ژنی CDKN2A(INK4A) و CDKN2B(INK4B) است که این ژنها P15 و P16 را کد میکنند و در بیش از 50% موارد T-ALL دیده میشود. موتاسیون در NOTCH1 که در بیشتر از 50% موارد وجود دارد، در اکثر مطالعات با پیشآگهی مطلوب همراه بوده یا هیچ تأثیری از پیشآگهی را نشان نداده است. فعالیت تیروزین کینازی غیر نرمال به صورت فیوژن ژنی NUP214(CAN)-ABL1) نیز در مواردی دیده شده و موتاسیون در JAK1 در 18% موارد دیده میشود و با پیشآگهی ضعیف همراه است (3).
علائم بالینی
اغلب موارد ALL بزرگسالان در ابتدا با علائم بالینی همراه هستند که حاصل از ناتوانی مغزاستخوان است (3). علائم فیزیکی مانند رنگ پریدگی، افزایش ضربان قلب، ضعف، خستگی ناشی از آنمی، پتشی و سایر اختلالات هموراژیک ناشی از ترومبوسیتوپنی و عوارض عفونی به علت نوتروپنی در بیماران دیده میشود (3،1). خونریزی یا عفونت در کودکان کمسنتر در حد شدیدتری دیده میشود (1). تب به علت عفونت یا سیتوکاینهای پیروژن مانند IL1,4 و TNFوجود دارد (1). درد استخوان یا مفصل به ندرت در ALL بزرگسالان دیده میشود، به طوری که زخمهای استخوانی در 1% موارد دیده میشود (3)، در حالی که ناهنجاریهای استخوانی مثل باندهای متافیزی، واکنش پریوستئال، استئولیز و استئواسکلروزیس در نیمی از کودکان دیده میشود (1). علائم بالینی در لوسمی ارتباط مستقیمی با انفیلتراسیون ارگانهای تیپیک با سلولهای بلاست دارد و از این موارد میتوان به لنفادنوپاتی، اسپلنومگالی و هپاتواسپلنومگالی اشاره کرد. انفیلتراسیون لوسمیک شبکیه، پوست، لوزهها، ریه و کلیهها مخصوصاً در موارد B-ALL و در موارد کمتری در T-ALL دیده میشود و در همه موارد با پیشآگهی ضعیف همراه است. درگیری طحال، کبد، تیموس، گرههای لنفاوی و CNS جزء موارد شایع میباشد. تودهی قدامی مدیاستن در موارد T-ALL دیده میشود (3،1).
علاوه بر موارد بالا درگیری maxilla، لق شدن دندان و التهاب لثه نیز در برخی موارد ALL کودکان دیده شده است، به طوری که در یک کودک 12 ساله با حضور سلولهای بلاست ALL در نمونه لثه همراه بوده است (4). فلج صورت که به صورت یک درگیری ثانویه در ALL دیده میشود نیز در مواردی گزارش شده است و استفاده از داروی پردنیزولون در درمان فلج صورت شاید بتواند به بهبود این عارضه کمک کند اما تشخیص لوسمی را به تأخیر میاندازد (8،7،6،5). در بیماران ALL به دلیل پیوند، ضعف سیستم ایمنی رخ میدهد و این افراد در معرض برخی عفونتهای باکتریایی مانند نوکاردیوز هستند که میتواند منجر به epididimo-orchitis و حتی آبسههای مغزی شود و گزارشاتی از حضور این علائم در بیماران ALL اهمیت این علائم در تشخیص لوسمی را بیان میکند (9).
مطالعات گذشته حضور آنتیژنهای تومور ویلمز (WT1) را در لوسمی ALL نشان داده است. بیان این آنتیژنها در ALL کودکان خیلی کمتر از بزرگسالان است و در حین عود سطح آن بالاتر از مراحل remission است. اخیراً دختری 3 ساله مبتلا به ALLبا حضور تومور ویلمز گزارش شده است.
ALL-EO (Pre B ALL with Eosinophilia) یک زیرگروه نادر از ALL میباشد که نسبت به لوسمی استاندارد به درمان مقاومتر است. t(5;14) در این لوسمی منجر به ائوزینوفیلی میشود و معمولاً با درگیری قلبی- تنفسی همراه است. موردی از لوسمی ALL-EO در یک پسر 13 ساله همراه با ناراحتی قلبی گزارش شده است و مصرف آسپارژیناز در این بیماران باید با احتیاط صورت گیرد و اکوکاردیوگرافی و MRI هرچه سریعتر باید انجام شود.
جدول شماره 2: علائم بالینی وآزمایشگاهی جدید در بیماران مبتلا به ALL در چند سال اخیر در ایران
| درگیری maxilla |
| لق شدن دندان |
| التهاب لثه |
| فلج صورت |
| Epididimo-orchitis |
| هیپر ائوزینوفیلی |
| تومور ویلمز |
| اختلالات قلبی-تنفسی در ALL-EO |
تشخیص آزمایشگاهی
شمارش اولیه لکوسیت از 0/1 تا /L10⁹ₓ1500 میباشد. شمارش بالای 10ₓ10⁹/L در بیش از نیمی از بیماران و شمارش بالای 100ₓ10⁹/L در 15-10% از بیماران دیده میشود. شمارش گرانولوسیتها اغلب 0.5ₓ10⁹/L˃ میباشد (نوتروپنی -0.5ₓ10⁹/L ˂- در 40% از بیماران دیده میشود). شمارش پلاکت اغلب ˃25ₓ10⁹/L و Hb کمتر از g/dl8 به ندرت دیده میشود. (برخلاف آن در کودکان معمولاً8g/dl ˂ میباشد) (3،1). هیپرائوزینوفیلی با حضور ائوزینوفیلهای غیرفعال ممکن است در حین تشخیص حضور داشته باشد و در حدود 44 بیمار در litretureها گزارش شده است (10). ائوزینوفیلی بیشتر در لام خون محیطی pre B-ALL دیده میشود و گزارش یک مورد از ائوزینوفیلی در پسربچه 5 ساله گواه این مطلب میباشد (12،11). کواگولوپاتی در حد متوسط در T-ALL دیده میشود و به ندرت با خونریزی شدید همراه است. در جمعیت کوچکی از بیماران، فیبرینوژن کمتر از 1g/dl است. DIC در موارد نادری دیده میشود (3،1). افزایش سطح اسید اوریک در نیمی از بیماران، هیپوکلسمی نادر و سطح فسفر و LDH نیز بالاست. در حدود بیش از 50% (یا حتی بیش از 90%) از سلولهای بلاست در مغز استخوان حضور دارند، تنها در کمتر از 15% موارد، سلولهای بلاست کمتر از 50% سلولهای هستهدار مغز استخوان را تشکیل میدهند. اغلب بیماران سلولهای بلاست در گردش دارند اما در مواردی که با شمارش پایین لنفوبلاستها همراه است (˂2ₓ10⁹/L) سلولهای بلاست در خون محیطی دیده نمیشود. آسپیراسیون یا بیوپسی مغز استخوان برای تشخیص اجباری است، تنها در کمتر از 15% بیماران، آسپیراسیون به علت خشک شدن مغز استخوان امکان پذیر نيست و باید از بیوپسی استفاده کرد. پونکسیون کمری برای بررسی درگیری CNS انجام میشود. اگر احتمال خونریزی به دلیل شمارش پایین پلاکت یا آلودگی به علت وجود تعداد زیاد بلاست در خون محیطی وجود دارد، باید پونکسیون را به تأخیر انداخت. درگیری CNS با حضور حداقل 5 لکوسیت در هر میکرولیتر CSF و حضور سلولهای بلاست یا فلج عصب جمجمه تأیید میشود، اما زمانی که تعداد لکوسیت در مایع نخاعی پایین است یا تشخیص مورفولوژی بلاست بینتیجه است بررسی ایمنولوژیک سلولهای بلاست درگیری CNS را تأیید میکند. درگیری CNS تقریباً در يك سوم ALL کودکان دیده میشود که اکثر این کودکان علامت نورولوژیکی خاصی ندارند و یک افزایش خطر برای عود محسوب میشود (3،1).
تشخیص افتراقی
علائم بالینی و آزمایشگاهی ALL میتواند با بسیاری از بیماریها اشتباه شود که افتراق آنها به تشخیص صحیح بیماری منجر میشود. افتراق ALL از لنفوسیتوز، لنفادنوپاتی و هپاتواسپلنومگالی در عفونتهای ویروسی و سایر لوسمیهای حاد و مزمن با بررسی مارکرهای سطحی، بررسی حضور یا عدم حضور لنفوسیتهای اتپیک و تیتر ویروس صورت میگیرد. بیماران با پرتوزیس و پاراپرتوزیس که لنفوسیتوز دارند نیز با ALL اشتباه میشوند که البته سلولهای درگیر در ALL لنفوبلاستها هستند. پتشی و اکموزیس و خونریزی حاد میتواند با ITP اشتباه شود که ITP اغلب با عفونت ویروسی، پلاکتهای درشت در اسمیر خون محیطی و عدم حضور آنمی همراه است. نوعی از ALL به نام Aleukemicpancytopenic ALL وجود دارد که با فقدان سلولهای بلاست در خون محیطی همراه است و باید از آنمی آپلاستیک افتراق داده شود و حتی میتواند یک سندرم پریلوسمیک باشد. برخلاف ALL، در آنمی آپلاستیک مغز استخوان هیپوسلولار است و ناهنجاری اسکلتی دیده میشود. در موارد نادری که ALL با انفیلتراسیون کم مغز استخوان همراه است، افتراق بین ALL و لنفوم غیرهوچکین لنفوبلاستیک معمولاً بر حسب درجه انفیلتراسیون که بیشتر یا کمتر از 25% باشد صورت میگیرد. درد استخوان، مفاصل و گاهی ورم مفاصل ممکن است الگوی رماتیسم مفصلی، تب روماتوئید، استئومیلیت یا سایر بیماریهای کلاژن را تقلید کند. بین تشخیص BCR-ABL (+)ALLاز CML در مرحلهی بلاستیک مشکل ایجاد میشود که در برخی موارد تشخیص نهایی تنها بعد از شروع درمان اولیه صورت میگیرد. در بیمارانی که بهبودي کامل مییابند شمارش خون محیطی مقادیر نرمال را نشان میدهد در حالی که در CML الگوی فاز مزمن (شیفت به چپ) را نشان میدهد. همچنین ALL کودکان باید از تومورهای گرد کوچک کودکان که در آن درگیری مغز استخوان مانند نوروبلاستوما، رابدومایوسارکوما و رتینوبلاستوما دیده میشود، افتراق داده شود (3،1).
پاتوفیزیولوژی
تغییرات ژنتیکی در مرکز پیشبرد سرطان خون هستند. بینظمی در ژنهایی که فاکتورهای نسخه برداری را کد میکنند باعث تخریب مسیرهای نسخه برداری- که هموستاز را در سلولهای هماتوپویتیک تنظیم میکنند- میشوند و مکانیزمی برای روند لوکموژنز فراهم میکنند. بینظمی در فعالیت تیروزین کینازها و جهش در گیرندههای آنها نیز هماتوپوئز را تحت تأثیر قرار میدهد. موتاسیون در FLT3 یکی از مثالهای آن است که این ژن یک رسپتور تیروزین کینازی را کد میکند و در سطح سلولهای هماتوپویتیک نابالغ بیان میشود و اثر سینرژیک با سایر فاکتور های رشد دارد. موتاسیون در NOTCH1– ژن کد کننده رسپتور گذار غشایی که رشد سلولهای T نرمال را فراهم میکند- معمولاً در T-ALL دیده میشود. این موتاسیون مسیر سیگنالینگ NOTCH1 را تحت تأثیر قرار میدهد و یک رویداد اولیـــــــــــــه در لوکموژنز میباشد. آنزیم گذار غشایی Y-secretase باعث مهار پیام رسانی مسیر NOTCH1 میشود و سنجش مهار کننده Y-secretase در موارد T-ALL انجام میشود. مطالعهای که اخیراً روی آنالیز SNP بر روی DNA، 242 مورد مبــــــــــتلا به B-precursor ALL کودکان انجام شده، آسیبهایی در ژنهای کد کنندهی تمایز سلولهای B در 40% موارد را نشان داده است. اکثر حذفها و جابجاییها دربرگیرنده ژن PAX5 بودند که تقریباً در يك سوم موارد باعث مسدود کردن تمایز پروژنیتورهای B نرمال به سمت بازآرایی زنجیره سنگین ایمنوگلوبولین میشود. سایر ژنهای جهش یافته که در ارتباط با بیشتر جابجاییها دیده شدهانـــــد شامل انکوژنهای کایمریک مثل TCF3-PBX1، ETV6-RUNX1، ژنهای لازم برای تکامل B-cell مثل TCF3، EBF، LEF1 و IKZF1(Ikaros) میباشد. مطالعات نشان داده که حذف IKZF در 7/83% از موارد ALL BCR-ABL(+) و در CML مرحله بلاستیک دیده شده است. حذف IKZF منجر به بیان منفی یا حذف کامل IKZF میشود و این امر در پیشرفت ALL BCR-ABL(+) نقش دارد. کمبود آنزیم GST (Glutathione S Transferase) با لوسمی نوزادان بدون بازآرایی MLL و ALL کودکان سیاه پوست همراه است. پلیمورفیسم NADPH با پیشرفت ALL همراه است. ژنوتیپ متغیر سایتوکروم P450 با افزایش خطر کودکان همراه است و گزارشی ارتباط بین پلیمورفیسم در اینترون 4 ژن α کتوردوکتاز 1C3 (AKR1C3) و پیشرفت لوسمی را نشان داده است، همچنین پلیمورفیسم در ژنهای مهارکننده چرخه سلول مانندCDKN2A و CDKN1B و CDKN2Bمیتواند منجر به لوکموژنز شود (1).
اتیولوژی
از مواردی که در ایجاد لوسمی مؤثرند میتوان به اشعه یونیزان و موتاژنهای شیمیایی اشاره کرد. علاوه بر آن در معرض قرار گرفتن در مقابل اشعه X در دوران جنینی نیز میتواند احتمال ابتلا به ALL را تا حدی افزایش دهد. صنعتی شدن، شرایط اقتصادی و انزوای اجتماعی با افزایش احتمال ابتلا به B-ALL کودکان همراه است. فقدان یا کاهش عفونت در اوایل زندگی ممکن است سیستم ایمنی بدن را به پاسخهای نابجا بعد از مواجهه با عوامل خارجی مستعد کند که به نوبه خود ممکن است باعث تسریع ALL از طریق تمایز یا استرس آپوپتوتیک شود. مواجه شدن با عوامل آسیب زننده DNA در دوران جنینی با لوسمی نوزادان با بازآرایی MLL همراه است (1).
علاوه بر موارد ذکر شده تحقیقات اخیر نشان داده که زندگی در نزدیکی خطوط برق فشار قوی ریسک ابتلا به ALL کودکان را بالا میبرد، به طوری که هر 600 متر فاصله از خطوط انتقال برق فشار قوی، 6/0 برابر ریسک ابتلا به ALL را میکاهد و در معرض قرار گرفتن در برابر بیش از یک خط انتقال برق فشار قوی، احتمال ابتلا را افزایش میدهد. میدان مغناطیسی نیز در ریسک ابتلا به ALL مؤثر است (13).
بررسی فاکتورهای پیشآگهی
چندین فاکتور که به عنوان فاکتورهای پیشآگهی دهنده طول مدت بقا در ALL شناخته شدهاند شامل سن، شمارش لکوسیتها، سطح LDH، کاریوتیپ، مرحله لوسمی در زمان پیوند استم سل، جنسیت دهندهی پیوند و سیکلوسپورین برای جلوگیری از GVHD میباشد (15،14).
مهمترین فاکتور، سن است. افزایش سن با کاهش نتیجه درمان همراه است. تعیین یک محدوده سنی برای تغییرات پیشآگهی لوسمی دشوار است اما تقریباً در همه مطالعات میزان LFS در بیماران بالای 60-50 سال پیشآگهی ضعیفی داشته است. بیماران بالای 60-50 سال با عوامل پرخطر که به بهبود کامل رسیدهاند و از لحاظ بالینی در موقعیت خوبی قرار دارند داوطلبان خوبی برای SCT میباشند. بیماران جوان (20-15 سال) بر طبق پروتکل درمانی ALL کودکان یا بر طبق پروتکل درمانی ALL بزرگسالان درمان میشوند (3). شمارش لکوسیت بالا در حین تشخیص (30-50ₓ10⁹/L˃) با پیشآگهی ضعیف همراه است. سن و شمارش لکوسیت در موارد B-ALL بکار میروند. در مورد B-ALL، pro B-ALL(t(4;11)) تقریباً در همه موارد جزء موارد پرخطر محسوب میشود و نیاز به دوز بالای سیتارابین و SCT میباشد. PH+ALL به عنوان ناخوشایندترین زیرگروه ALL بزرگسالان محسوب میشود و میزان وقوع آن با افزایش سن بالا میرود. درگیری CNS بیشــتر در موارد B-ALL دیده میشود. بارزترین پیشآگهی در T-ALL نوع زیرگــــــــروه میباشد، به طوری که در موارد early T-ALL، thymic T-ALL و mature T-ALL به ترتیب با میزان LFS 25%، 63%، و 28% همراه است. موارد thymic T-ALL پاسخ درمانی ضعیف و احتمال عود بالایی دارند. افزایش بیان HOX1، HOX11L2، SIL، TAL1 و CALM-AF10 با زیرگروههای تیموس همراه است. با رژیمهای درمانی اخیر میزان بهبود کامل به بیش از 90% و میزان LFS به 60-40% در موارد T-ALL رسیده است (3).
تغییرات ژنتیکی در برخی ژنها پاسخ به درمان در ALL را میتواند تغییر دهد. مطالعات نشان داده که افزایش بیان MRP1 -پروتئین گذار غشایی را کد میکند که متعلق به خانواده C در ABC-transporterها میباشد-در بیماران ALL با مقاومت داروئی و عود دیده شده است. علاوه بر آن حضور برخی پلیمورفیسمها در ژن MRP1 در بیان ژن مزبور تأثیرگذار است، اما مطالعهای که در ایران انجام شد افزایش بیان این ژن بدون حضور پلیمورفیسم بوده است (16).
حضور یا عدم حضورآنتیژنهای میلوئیدی در میزان بقا و طول دوره بهبود کامل تأثیر چشمگیری دارد؛ زمانی که بیان آنتیژنهای میلوئید در سطح سلولهای بلاست وجود دارد میزان بقا و طول دوره بهبود کامل به طرز چشمگیری کاهش مییابد (17).
BCL2 یک فاکتور آنتیآپوپتوتیک مهم است. بررسی بیان BCL2 در سطح بلاستهای ALL کودکان نشان داده است که بین بیان یا عدم بیان BCL2 با میزان بقا و طول دوره بهبود کامل در بیماران ALL ارتباط معناداری وجود ندارد (17)، اما مطالعاتی که بر روی ALL بزرگسالان انجام شده نشان داده که بیان BCL2 در سطح سلولهای بلاست ALL با نتایج بالینی نامطلوب همراه است (18). این تفاوت بین نقش BCL2 در ALL کودکان و بزرگسالان نشان میدهد که ALL در کودکان و بزرگسالان بیماریي مجزا بر اساس ساختارهای مولکولی مخصوص به خود میباشد (17).
مطالعات گذشته نشان داده که پلیمورفیسم در آنزیمهایی که در متابولیسم فولات نقش دارند نیز در ریسک ابتلا به ALL نقش دارد، اما مطالعهای که در غرب ایران انجام شده دو پلیمورفیسم TS28-bp repeat(در آنزیم تیمیدین سنتتاز) و MS A2756G (در آنزیم متیونین سنتتاز) را مورد بررسی قرار داده و نتایج نشان داد که حضور هیچ کدام از این پلیمورفیسمها تأثیری در ریسک ابتلا به ALL ندارد (19). علاوه بر آن بررسی پلیمورفیسمهای C677T و A1298C در جمعیت غرب ایران ارتباطی با افزایش ریسک ALL نشان نداده است.
مطالعهای که بر روی CD مارکرهای دخیل در تمایز پروکرسورهای سلول B (شامل CD27,CD44) صورت گرفته نشان داد که در زمان بیان CD27، سطح لکوسیت پایین، تعداد بلاست در مغز استخوان پایین و پلاکت بالا بود و بیماران CR و بقاي طولانیتری داشتند و CD27 اثر CD44 را که با لکوسیت و بلاست زیاد در مغز استخوان همراه است را تعدیل میکند، به طوری که بیمارانی که برای CD27/CD44 مثبت بودند نزدیک به گروه CD27 مثبت بودند.
HLAها نیز در استعداد ابتلا به ALLدخیل هستند، به طوری که HLA-DQB1 افزایش معنیداری در ALL کودکان و بزرگسالان داشته است. HLA-DQ5 به عنوان یک استعداد اللیک برای ALL محسوب میشود و مشاهده پروفایل اللیک افزایش HLA-DQ7 و کاهش HLA-DQ2 در کودکان ALL با کاهش بقا در این کودکان همراه است.
اختلالات انعقادی و ترومبوز از دیگر مشکلات بیماران ALL است. بررسی پروتئینهای ضد انعقادی در این بیماران کاهش سطح پروتئین C و آنتیترومبین و افزایش سطح پروتئین S و Dدایمر را نشان داده است و بیماران با این پروفایل و شمارش لکوسیت بالا و پلاکت پایین باید برای ترومبوز و DIC بعد از شروع یا در طول شیمی درمانی پیگیری شوند و بروز ترومبوز به دنبال مصرف آسپارژیناز و استروئید به تغییر در سطح این پروتئینهای انعقادی نسبت داده شده است.
بررسی حضور ویروس انکوژنیک BK؛ ویروس در کودکانی که تازه برای ALL تشخیص داده شدهاند نشان داد که حضور این ویروس در ادرار این کودکان غیر معمول نیست ولی ارتباط معناداری با تناوب عود در این بیماران ندارد.
جدول شماره 3: تأثیر فاکتورهای پیشآگهی بررسی شده در بیماران مبتلا به ALL در سالهای اخیر در ایران
| نوع تأثیر مربوطه | متغیر بررسی شده |
| افزایش بیانMRP-1 با افزایش مقاومت دارویی و عود | بیان MRP-1 |
| کاهش بقا و طول دوره بهبود کامل | بیان آنتیژنهای میلوئید |
| عدم تأثیر بر میزان بقا و طول دوره بهبود کامل | بیان BCL-2 در ALL کودکان |
| نتایج بالینی نامطلوب در میزان بقا و طول دوره بهبود کامل | بیان BCL-2 در ALL بزرگسالان |
| عدم تأثیر در ریسک ابتلا به ALL | حضور پلیمورفیسم در آنزیمهای دخیل در متابولیسم فولات |
| کاهش پروتئین C و آنتیترومبین و افزایش پروتئین S و Dدایمر با افزایش خطر ترومبوز همراه است | پروتئینهای ضد انعقادی شامل پروتئینc و s، آنتیترومبین و Dدایمر |
| حضور BK ویروس در ادرار و عدم ارتباط آن با عود متناوب | حضور BK ویروس در ادرار کودکان ALL و ارتباط آن با عود متناوب |
| حضور CD27 با بهبود کامل بالاتر و بقا طولانیتر همراه است در مقایسه با موارد حضور CD44 | CDهای دخیل در تمایز پروکرسورهای سلول B (CD27,CD44) |
| HLA-BQ5 به عنــوان یک استعداد اللیک ALL، افزایش HLA-DQ7 و کاهش HLA-DQ2 همراه با کاهش بقا در کودکان ALL | HLA-DQB1 در ALL کودکان و بزرگسالان |
درمان
درمان لوسمی لنفوبلاستیک حاد در موارد کودکان و بزرگسالان تفاوتهای زیادی ندارد، به طوری که در درمان اطفال تمرکز اصلی در بهینه سازی رژیمهای شیمی درمانی فشرده میباشد، در حالی که در بزرگسالان روی درمان با دوز بالا مانند موارد AML و استفاده از پیوند مغز استخوان تمرکز میکنند؛ علاوه بر آن پایبندی به پروتکلها و تشویق بیماران به انطباق بیشتر آنها در مشاوره روانی در ALL بزرگسالان امری ضروری است (3).
با شیمی درمانیهای جدید که در موارد ALL صورت میگیرد میزان بقاي 5 ساله از 75% به 87% رسیده است. رویکرد کلی درمان متکی به سه فاز اصلی میباشد:
– Remission induction therapy
– Intensification (consolidation) therapy
– Continuing therapy (1)
Remission induction therapy شامل گلیکوکورتیکوئیدها (پردنیزون، پردنیزولون و دگزامتازون)، وینکریستین و آسپارژیناز با یا بدون داروهای دیگر مثل آنتراسایکلین و سیکلوسفامید میباشد. تشدید این نوع درمان اگرچه باعث اثربخشی بیشتر از طریق حذف سلولهای بلاست میشود، اما در بیماران با عدم حضور فاکتورهای پیشآگهی نامطلوب لازم نيست. تشدید این نوع درمان با بروز عوارض زود هنگام و افزایش مرگ و میر همراه است. در بین گلیکوکورتیکوئیدها دگزامتازون به دلیل نیمه عمر بالا و افزایش نفوذ به CSF نسبت به پردنیزون و پردنیزولون اثربخشتر است. علاوه بر نتایج مطلوب در برخی مطالعات مصرف آنها با عفونتهای تهدید کننده و سپتیک کشنده همراه است. دوز مصرفی این داروها نیز در نتایج حاصل از درمان مؤثر است. اکثر رژیمهای درمانی در این نوع درمان شامل آسپارژیناز میباشد، با این حال استفاده ار آن به علت عوارض جانبی در حال محدود شدن است (1).
Intensification (consolidation) therapy: معمولاً در بیمارانی که بهremission میرسند کاربرد دارد و در بیماران با ریسک بالا و آنهایی که به درمان القایی اولیه پاسخ آهسته میدهند مفید است. داروهایی که در این نوع درمان استفاده میشود مشابه درمان قبلی میباشد و بیماران با زیرگروههای مختلف پاسخ خوبی به این نوع درمان میدهند؛ برای مثال بیماران با ETV6-RUNX با گلوکورتیکوئید، وینکریستین و آسپارژیناز نتایج مطلوبی نشان دادهاند، همچنين دوز بالای متوتروکسات (g/m²5) در موارد T-ALL با نتایج مطلوب همراه بوده است (1).
Delayed intensification therapy یا reinduction که به طرز گستردهای استفاده میشود و شامل تکرار یا تشدید remission induction سه ماه بعد از پایان درمان القایی میباشد، در درجه اول به نفع افرادی است که در معرض عود بالا قرار دارند (1).
Continuation therapy: به جز بیماران مبتلا به B-ALL (بیماران B-ALL نیاز به دوز بالای متوتروکسات، سیتارابین و سیکلوفسفامید دارند و درمان نگهدارنده در آنها حذف شده است)، همهی بیماران نیاز به این نوع درمان دارند که به طور معمول 5/2-2 سال طول میکشد. کوتاه کردن این زمان منجر به نتایج نامطلوب میشود. این نوع درمان نسبت به درمان با ایجاد وقفه و استفاده از دوز بالای داروهای شیمی درمانی، مؤثرتر است. از ترکیبات رایج در این نوع درمان استفاده از متوتروکسات و مرکاپتوپورین به طور هفتگی یا روزانه میباشد. در برخی مواقع عوارض ناشی از این داروها منجر به قطع یا کاهش دوز دارو میشود. مصرف مرکاپتوپورین به صورت خوراکی نسبت به نوع تزریقی بالاترین اثر را دارد. در رابطه با متوتروکسات کاهش جذب در فرم تزریقی و انطباق ضعیف با فرم خوراکی آن از مشکلات مصرف این دارو است. در برخی مطالعات تیوگوانین در مقایسه با مرکاپتوپورین اثر آنتیلوکمیک قویتری داشته است اما مصرف آن با دوز بالای mg/m²40 به صورت روزانه با افزایش توکسیسیته همراه بوده است. درمان فشرده با مرکاپتوپورین و درمان نگهدارنده با متوتروکسات مربوط به حضور بدخیمی ثانویه مخصوصاً در بیماران با کمبود تیوپورین متیل ترانسفراز میباشد. مطالعات نشان دادهاند که ترکیب وینکریستین و گلوکوکورتیکوئید با مرکاپتوپورین/متوتروکسات مفید است (1).
استفاده از مهار کنندههای تیروزین کیناز ABL (ایماتینیب/گلیوک) در موارد PH(+)ALL کاربرد دارد. ترکیب ایماتینیب با شیمی درمانی باعث میزان بهبودي تا 90% و ترمیم مولکولی در 50% موارد میشود. مطالعات نشان داده است که اکثریت بیماران با حضور MRD بعد از پیوند با مصرف ایماتینیب بقاي طولانیتری دارند. در بیماران مسن که نمیتوانند تحت پیوند استم سل قرار بگیرند و نتایج ناخوشایندی از شیمی درمانی نشان دادهاند، ایماتینیب به عنوان تک داروی درمانی محسوب میشود. نسل دوم تیروزین کینازهـــــــا (Dasatinib، Nilotinib) در مقایسه با ایماتینیب اثربخشی بیشتری دارند و در موارد موتاسیون در گیرنده تیروزین کینازها (به جز موتاسیون T3151) فعالند و میزان بهبودی در بیمارانی که از این داروها استفاده میکنند 30% است. یک علت عود در بیمارانی که ایماتینیب مصرف میکنند به دلیل جهش در گیرنده تیروزین کینازها و ایجاد مقاومت داروئی میباشد، بنابراین قبل از درمان عود بايد آنالیز مغز استخوان برای بررسی مقاومت علیه دارو انجام شود (3).
استفاده از Abهای منوکلونال یک راه درمانی مهم در ALL بزرگسالان است و ترکیب آنها با شیمی درمانی امیدوار کننده میباشـــــــــــد. استفاده از Anti-CD20 در درمان mature B-ALL موفق بوده و در موارد B precursor ALL نیز تحت بررسی قرار دارد (3).
داروی متوتروکسات که یک متابولیت ضد فولات است در موارد بدخیمی و غیر بدخیمی کاربرد دارد. یکی از موارد کاربرد این دارو در لوسمی ALL است (22،21،20).
آنتیبیوتیک ونکومایسین در موارد عفونتهای باکتریایی در بیماران ALL کاربرد دارد. امروزه مقاومت به ونکومایسین (VRE) در جمعیتهای مختلف دیده شده و میزان و علت شیوع آن در جمعیتهای مختلف متفاوت است. مطالعهای که در روی کودکان مبتلا به ALL در ایران انجام شده نشان داده که شیوع VRE در این کودکان بالاست و اغلب موارد مثبت برای VRE ژنوتیپ vanA را نشان میدهند و مطالعات گستردهتری برای تشخیص ریسک فاکتورهای ایجاد VRE در این بیماران لازم است (26).
تعدادی از داروهای جدید نیز در درمان ALL تحت بررسی قرار دارند که از آنها میتوان به آنالوگهای پورین و داروهای مرتبط مثل Nelarabine، Clofarabine، مهار کنندههای کیناز و لیپوزومهای تهیه شده از وینکریستین و سیتارابین برای استفاده داخل نخاعی اشاره کرد (3).
استفاده از پروستاگلاندین E2(PG E2) برای اولین بار بر روی رده سلولی ALL بررسی شد و اثرات ضد تکثیری و آپوپتوزی این دارو با ED50 قابل پذیرش و تأیید شد، همچنین داروی مزبور باعث القاي تمایز نهایی بلاستهای ALL از طریق بلوکه کردن چرخه سلول در فاز G0/1 شد. یافتههای مزبور میتواند به طراحی پروتکل درمانی جدید در آینده کمک کند.
تأثیر یک ترکیب غیر نوکلئوزیدی به نام BIBR1532 به عنـــوان یک داروی ضد سرطان بر روی رده سلولی Pre B ALL بررسی شد. غلظت بالای این دارو از طریق سرکوب survivin (عضوی از خانواده مهار کنندههای آپوپتوز) منجر به کاهش فعالیت تلومراز و سرکوب چرخه سلول میشود كه میتواند به عنوان داروی ضد سرطان مورد استفاده قرار بگیرد.
پیوند استم سل آلوژنیک (SCT)
SCT در بیماران بزرگسال پرخطر نتایج مطلوبی را نشان داده است. در بیماران جوانتر برای مثال در کودکانی که میزان بقا با شیمی درمانی به تنهایی پیشرفت ميکند، میزان SCT کاهش مییابد، با این حال انجام SCT نیاز به بهینه سازی دارد که شامل در دسترس بودن دهنده مناسب، فاکتورهای مربوط به بیمار، مارکرهای بیماری، پاسخ به درمان و در دســـــــترس بودن داروها میباشد (3). بیماران با فیوژن ژنی BCR-ABL1 و ETP ALL، که میزان عود در آنها سریعتر و پاسخ به درمان ضعیفتر است کاندیداهای بیشتری برای پیوند هستند. استفاده از ایماتینیب و سایر مهار کنندههـــــــای تیروزین کیناز که منجر به بهبود پاسخ در درمان اولیه در بیماران BCR-ABL1 میشوند این سؤال را ايجاد ميكند که آیا در این موارد پیوند در اولین بهبود توصیه میشود یا خیر (1). تاکنون پیوند استم سل در زیرگروههای پرخطر شامل نوزادان و مواردی که با بازآرایی MLL همراهند، نتایج مطلوبی را نشان نداده است. میزان MRD بعد از پیوند، خطر عود بعد از پیوند را پیشبینی میکند. برای مثال در بیماران T-ALL، سطح MRD بالا قبل از پیوند (1-1/0%) با عود بالا بعد از پیوند همراه است و بیماران با سطح MRD کمتر میزان بقاي 2 ساله 50-30% و بیماران با MRD منفی میزان بقاي 2 ساله 70% دارند (1).
گستردهترین رژیم درمانی برای پیوند Hsc آلوژن، اشعه دادن کل بدن (Total Body Irradiation=TBI) میباشد. استراتژی های جدید در حال حرکت به سمت استفاده از رژیمهای شیمی درمانی مناسب به عنوان جایگزینی مناسب برای اشعه درمانی و جلوگیری از عوارض ناشی از آن است. در مطالعهای که از رژیمهای غیر از اشعه درمانی (استفاده ترکیبی از بوسولفان و سیکلوفسفامید به صورت ترکیبی قبل از پیوند) در کودکان مبتلا به ALL انجام شد، مشاهده گرديد که میزان بقا در بیماران رضایت بخش بوده است.
یکی از مشکلاتی که بعد از BMT با آن مواجه هستیم، GVHD حاد و مزمن میباشد. میانگین زمان وقوع GVHD حاد در بیماران ALL، 3/13 روز و GVHD مزمن 123 روز میباشد. رژیم دارویی BUCY (بوسولفان + سیکلوسفامید) به همراه تزریق مرتب از سلولهای BM دهنده، به عنوان درمان مناسب در بیماران ALL برای جلوگیری از پروفیلاکسی GVHD میباشد. در بیماران ALL احتمال خطر در 6 ماههی اول بعد از SCT بیشتر از احتمال خطر در 6 ماههی دوم بعد از پیوند میباشد (27).انتخاب یک مدل آماری مناسب برای تشخیص دقیق و قابل اعتماد فاکتورهای پیشآگهی ضرورت دارد. با استفاده از مدلهای آماری پارامتریک و غیر پارامتریک میتوان میزان بقاي بعد SCT را پیشبینی کرد (28). در مطالعهای که با استفاده از آمار پارامتریک انجام شده و در آن برای اولین بار در ایران از مدل آماری Generalized Gamma-که جزء مدلهای آماری پارامتریک میباشد- استفاده گرديد، مزیت استفاده از مدلهای آماری پارامتریک در پیشبینی میزان بقا در بیماران ALL که CST داشتهاند، نشان داده ميشود. بین سن اهدا کننده و شمارش WBC، سطح CD3، GVHD حاد و مزمن و جبران پلاکتی با میزان بقا بعد از BMT ارتباط وجود دارد (28،27). به ازای هر 1000 عدد افزایش تعداد WBC، میزان بقا 6% افزایش مییابد و به ازای هر واحد اضافه شدن به CD3 میزان بقا 6/4% افزایش مییابد. همچنین در بیمارانی که به دنبال SCT جبران پلاکتی دارند میزان بقا 9/3 مرتبه نسبت به بیمارانی که جبران پلاکتی نداشتند طولانیتر میباشد. علاوه بر آن تعدیل برخی فاکتورهای پیشآگهی در پیشبینی میزان بقا مؤثر است. در بیماران بدون عود با تعدیل سن و جنس میزان بقا 10 مرتبه طولانیتر است. CGVHD و جبران پلاکتی تعدیل شده برای سن و جنس ارتباط معناداری با میزان بقا دارد اما AGVHD تعدیل شده برای سن و جنس ارتباط معناداری ندارد. CGVHD تعدیل شده برای AGVHD و جبران پلاکتی و تعداد WBC تعدیل شده برای سن و جنس ارتباط معناداری با میزان بقا بعد از SCT ندارد. جبران پلاکتی تعدیل شده برای CGVHD و AGVHD ارتباط معناداری با میزان بقا دارد (27).
همچنین با استفاده از مدلهای آماری احتمال مرگ بعد از SCT را نیز میتوان تخمین زد. احتمال مرگ در بیمارانی که جبران پلاکتی ندارند 14/2 برابر بیمارانی است که جبران پلاکتی دارند و در بیمارانی که AGVHD پیشرفته دارند حدود 14/2 برابر بیمارانی است که AGVHD ندارند، این تخمین در شرایط تعدیل جبران پلاکتی، CGVHD، سن و جنس بیماران میباشد (27).
جدول شماره 4: انواع درمان در ALL
| توضیحات | نوع درمان |
| شامل گلیکوکورتیکوئیدها (پردنیزون، پردنیزولون و دگزامتازون)، وینکریستین، آسپارژیناز با یا بدون داروهای دیگر مثل آنتراسایکلین و سیکلوسفامید
نتایج حاصل از این نوع درمان در مطالعات مختلف متغیر بوده است. |
Remission induction therapy
|
| داروهای آن مشابه با درمان remission induction therapy است.
کاربرد این نوع درمان در مرحلهremission است. |
Intensification (consolidation) therapy |
| همه بیماران به جز بیماران مبتلا به B-ALL به مدت 5/2-2 سال تحت این نوع درمان قرار میگیرند.
داروهای رایج در این نوع درمان مرکاپتوپورین و متوتروکسات میباشد. |
Continuation therapy
|
| Anti CD20 در موارد mature B-ALL | آنتیبادیهای منوکلونال |
| آنالوگهای پورین، مهارکنندههای کیناز، لیپوزومهای تهیه شده از وینکریستین و سیتارابین | سایر داروها |
| در بزرگسالان در گروه پرخطر نتایج مطلوبی را نشان داده است. | پیوند استم سل آلوژنیک |
پیشگیری و درمان لوسمی CNS
انفیلتراسیون مننژ با سلولهای لوسمیک موجب میشود سد خونی- مغزی به عنوان محافظ برای سلولهای بلاست عمل کند و مانع اثر درمانهای سیستمیک روی سلولهای بلاست شود. از سال 1970 رادیوتراپی جمجمه (14-12Gy) و متوتروکسات بعد از Remission Induction Therapy به عنوان اقدامی پیشگیرانه برای جلوگیری از لوسمی CNS بوده و در درمان ALL کودکان کلیدی است. رادیوتراپی اثرات جانبی دارد و کاهش دوز اشعه به کمترین حد آن حفاظت کافی در برابر عود CNS میدهد. حذف کامل رادیوتراپی جمجمه میزان قابل قبولی از عود را نشان داد اما میزان بقا در بیماران کاهش یافت. درمان فشرده و منظم داخل نخاعی بدون رادیوتراپی جمجمه برای پیشگیری از لوسمی CNS در نوزادان با تشخیص لوسمی CNS کافی است. درمان منظم شامل دوز بالای متوتروکسات، آسپارژیناز با دوز تشدید شده و دگزامتازون بعلاوه درمان داخل نخاعی برای کنترل لوسمی CNS لازم میباشد. درمان سه گانه با متوتروکسات، سیتارابین و هیدروکوتیزون نسبت به درمان داخل نخاعی تنها با متوتروکسات در جلوگیری از عود CNS مؤثرتر است. در موارد حضور سلولهای بلاست در CSF درمان داخل نخاعی باید تشدید شود؛ چون این یافته به عنوان یافتهای برای افزایش احتمال عود CNS و نتایج بالینی نامطلوب میباشد (1).
MRD
پاسخ به درمان، که با بررسی مرفولوژی سلولهای مغز استخوان و اسمیر خون محیطی انجام میشود، حساسیت و صحـــــــــــت کار را كاهش ميدهد. ظهور روش (تشخیص حداقل بیماری باقیـــــــــــــــمانده) MRD (Minimal Residual Disease)، حداقل 100 برابر حساستر از تکنیکهای مرفولوژیکی معمولی میباشد و یک راه جدید نظارت بر درمان را معرفی کرده است. از روشهای قابل اعتماد برای اندازهگیری MRD میتوان به، flow cytometric profiling of aberrant immunophenotypes,
PCR amplification of fusion transcripts and chromosomal breakpoints, and PCR amplification of antigen – receptor genes اشاره کرد. بیمارانی که MRD آنها در طول یا انتهای Remission Induction Therapy در مغز استخوان 01/0% یا بیشتر باشد احتمال عود به مراتب بالاتری دارند، در حالی که افرادی که MRD آنها در پایان Remission Induction Therapy 1% یا بیشتر باشد و افراد با MRD 1/0% یا بیشتر در طول Continuation Therapy خطر عود بسیار بالایی دارند (1).
عوارض درمان
پیشرفت در درمانهای محافظت کننده میزان مرگ و میر را به کمتر از 2% کاهش داده است و میزان بقاي 5 ساله از صفر به 80% رسیده است (29). به دنبال افزایش میزان بقا، اثرات جانبی درمان مانند نقص در ترشح هورمون رشد و هورمونهای تیروئیدی و سندرمهای متابولیک ظاهر میشود (31،30). در مطالعهای که در سالهای اخیر انجام شده شیوع سندرم متابولیک در کودکان بازمانده طولانی مدت بعد از درمان ALL بالا بوده است. این سندرم در فاصلهای نزدیک بعد از درمان کامل ALL ظاهر شد و تشخیص زود هنگام این سندرم میتواند احتمال خطر بیماریهای کاردیوواسکولار و کشندگی را کاهش دهد. همچنین کنترل وزن و انجام تمرینهای فیزیکی در پیگیری این بیماران توصیه میشود (32).
درمانهای remission induction شامل پردنیزولون، وینکریستین و آسپارژیناز در 20-10% از بیماران منجر به هیپرگلیسمی میشود، مخصوصاً در بزرگسالان و بیماران با فربهی و تاریخچه خانوادگی از دیابت ملیتوس یا سندرم داون. علاوه بر آن در 5-3% بیماران منجر به مشکلات انعقادی مثل ترومبوز پیش رونده میشود. تشدید استفاده از متوتروکسات و گلوکوکورتیکوئیدها با افزایش نوروتوکسیسیتی و استئونکروز همراه است. انباشته شدن زیاد آنتراسایکلین باعث کاردیومیوپاتی شدید مخصوصاً در کودکان کم سن میشود. رادیوتراپی جمجمه باعث نئوپلاسم ثانویه، اختلال نوروفیزیولوژیک و ناهنجاری اندوکرین، فربهی، بلوغ زودرس، کوتاهی قد و پوکی استخوان میشود (1).
از عوارض جانبی آسپارژیناز میتوان به تغییرات هماتولوژیک، هیپرگلیسمی، واکنشهای آلرژیک، آنسفالوپاتی، درد شکم، پانکراتیت، اختلال عملکرد کبد، عوارض گوارشی انعقادی و ترومبوز اشاره کرد. همچنین مطالعات اخیر نشان داده که سطح سرمی TG به دنبال مصرف آسپارژیناز کاهش مییابد، کلسترول تغییر معنیداری پیدا نمیکند وLDL افزایش مییابد (35،34،33). به طور کلی آسپارژیناز با دوز 6000 واحد در متر مربع برخلاف دوزهای بالای آن باعث افزایش چربیهای خون نمیشود (36).
آسیب کلیوی یکی از عوارض مهم متوتروکسات میباشد (24،23)؛ اما مطالعهای که بر روی بیماران مبتلا به ALL انجام شده نشان داد که شیوع آسیب کلیوی به دنبال درمان با دوز بالای متوتروکسات (دوز بالاتر از g/m²1) كم است و همهی آسیبها به فاز اول و دوم آسیب کلیوی محدود میشوند و بیماران خودبخود بهبود مییابند (25).
عود
با وجود اینکه میزان بهبود کامل در کودکان ایرانی مبتلا به ALL، 79% است (37)، هنوز 30-25% از این بیماران با عود مواجه هستند (38) و حداقل 15% از این بیماران نیز فوت میکنند. عود یک علت شایع شکست درمانی در ALL کودکان است (39).
عود معمولاً در طول درمان یا بعد از درمان، 2 سال بعد از قطع درمان یا حتی 10 سال بعد از تشخیص نیز بروز میکند. مغز استخوان به عنوان شایعترین محل درگیر در عود میباشد و میزان عود در محلهای خارج از مغز استخوان مانند CNS و بیضهها کاهش یافته است. عود مغز استخوان با یا بدون درگیری نقاط خارج از مغز استخوان با پیشآگهی ضعیف همراه است. بیماراني که تنها عود در مغز استخوان دارند نسبت به بیماراني که علاوه بر مغز استخوان درگیری نقاط دیگر را هم دارند، با وضعیت بدتری مواجهند. در مقابل بیمارانی که عود CNS دارند پیشآگهی بهتری دارند. در کودکان مبتلا به ALL همراه با عود، عوامل خطر شامل بهبود اولیه کوتاه مدت، ایمنوفنوتیپ سلول T، ALL BCR-ABL(+)، حضور سلولهای بلاست در گردش یا شمارش لکوسیت بالا در عود میباشد. حضور MRD در Remission Induction دوم یک فاکتور پیشآگهی دهنده نامطلوب میباشد، اگرچه ممکن است شیمی درمانی برای بهبود ثانویه طولانی مدت در بیماران بدون علائم پرخطر کافی باشد اما پیوند استم سل آلوژنیک گزینهای مناسب برای بیماران باقی مانده مخصوصاً آنهایی که دچار عود خونی در طی درمان و یا مدت کوتاهی پس از آن میشوند یا آنهایی که با فنوتیپ T-ALL همراهند و آنهایی که MRD دارند، میباشد (1).
یک علت اصلی عود، مقاومت به معرفهای آنتیلوکمیک است. گلیکوکورتیکوئیدها یکی از درمانهای اصلی در ALL محسوب میشوند و در شیمی درمانیهای چند داروئی در ALL کودکان کاربرد دارند (40). در برخی مطالعات گذشته نشان داده شده که برخی پلیمورفیسمها در رسپتور گلیکوکورتیکوئیدها (شامل BCL1,N363s,ER22/23EK) میتواند باعث افزایش یا کاهش حساسیت به گلیکوکورتیکوئیدها و عود شود (41-46)؛ اما مطالعهای که در ایران انجام شد نشان داد كه شیوع پلیمورفیسم مزبور در جمعیت مورد مطالعه پایین است و ارتباطی با احتمال عود در ALL ندارد (47).
References:
- Campana D, PuiCh-H. Childhood acute lymphoblastic leukaemia. In: Hoffbrand AV, Catovsky D, Tuddenham GD E, Green A R, editors. Postgraduate Haematology. 6th ed. Blackwell: John Wiley & Sons Ltd; 2010. P.448-60.
- Mousavi SM, Gouya MM, Ramazani R, Davanlou M, Hajsadeghi N, Seddighi Z. Cancer incidence and mortality in Iran. Ann Oncol 2009;20:556-63.
- Gökbuget N, Hoelzer D.Adult acute lymphoblastic leukaemia. In: Hoffbrand AV, Catovsky D, Tuddenham GD E, Green A R, editors. Postgraduate Haematology. 6th ed. Blackwell: John Wiley & Sons Ltd; 2010.P.433-46.
- FallahinejadGhajari M, Moshref M, Taghipour. Maxilla Unilateral Swelling as the First Diagnostic Symptom of Acute Lymphoblastic Leukemia Relapse: A Case Report. J Dent (Tehran) 2011;8(1):44-47.
- Karimi M, Cohan N, Zareifar S, Inaloo S, Kalikias S, Moslemi. Initialpresentation of childhoodleukaemia with facialpalsy: threecasereports. BMJ Case Rep 2009;2009.
- Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med 2004;350: 1535–48.
7.Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med 1999; 341: 1051–62.
- Bilavsky E, Scheuerman O, Marcus N, Hoffer V, Garty BZ. Facial paralysis as a presenting symptom of leukemia.PediatrNeurol 2006;34:502–4.
- Dehghani M, Davarpanah MA. Epididymo-Orchitis and Central Nervous System Nocardiosis in a Bone Marrow Transplant Recipient for Acute Lymphoblastic Leukemia.Exprimental and Clinical Transplantation 2009;7(4):264-66.
- D’Angelo G, Hotz AM, Todeschin P. Acute lymphoblastic leukemia with hypereosinophilia
and 9p21 deletion: case report and review of the literature. Lab Hematol 2008;14(1):7-9.
- Wilson F, Tefferi A. Acute Lymphocytic Leukemia with Eosinophilia; Two case reportus
and a literature review. LeukLymphorna 2005;46(7): 1045-50.
- قریشی ضیاءالدین، رضامند عظیم. گزارش یک مورد لوسمی لنفوبلاستیک حاد در کودک 5 ساله با ائوزینوفیلی. مجله پزشکی ارومیه 1389؛21(5):440-443.
- Sohrabi MR, Tarjoman T, Abadi A, Yavari P. Living Near Overhead High Voltage Transmission Power Lines as a Risk Factor for Childhood Acute Lymphoblastic Leukemia: a Case-control Study. Asian Pacific J Cancer Prev 2010;11:423-27.
- Le QH, Thomas X, Ecochard R, Iwaz J, Lheritier V, Michallet M, et al. Initial and late prognostic factors to predict survival in adult acute lymphoblastic leukaemia 3.Eur J Haematol 2006;77:471-479.
- Silverman LB. Acute lymphoblastic leukemia in infancy.Pediatr Blood Cancer 2007; 49:1070-1073.
- Mahjoubi F, Akbari S, Montazeri M, Moshyri F. MRP1 polymorphisms (T2684C, C2007T, C2012T, and C2665T) are not associated with multidrug resistance in leukemic patients. Genet Mol Res 2008;7(4):1369-74.
- Amirghofran Z, Daneshbod Y, Gholijani N, Esmaeilbeing.The Infuence of Bcl-2 and Myeloid Antigen Expression on Response to Therapy in Childhood Acute Lymphoblastic Leukemia. Arch Iran Med 2011;14(3):170-174.
- Amirghofran Z, Daneshbod Y, Gholijani N. BCL-2 in combination to myeloid antigen expression in adult acute lymphoblastic leukemia and prognostic outcome.OncolRes2009;17:447 –54.
- Rahimi Z, Ahmadian Z, Akramipour R, Madani H, Mozafari H, Vaisi-Raygani A. Thymidilate Synthase and Methionine Synthase Polymorphisms in Children with Acute Lymphoblastic Leukemia in Western Iran. International journal of hematology-oncology and stem cell research 2010;39(3):2195-200.
- Jolivet J, Cowan KH, Curt GA, Clendeninn NJ, Chabner BA. The pharmacology and clinical use of methotrexate. N Engl J Med 1983;309:1094-104.
- Oguz A, HasanOglu A, Azgu FS, Timlioglu O, Biberoglu G, Uluoglu C. Methotrexate related hepatotoxicity. Gazi Med J 2002;13:69-72.
- Hersh EM, Wong VG, Henderson ES, Freireich EJ. Hepatotoxic effect of methotrexate. Cancer 1966;4:600-6.
- Lawrenz-Wolf B, Wolfrom C, Frickel C, Fengler R, Wehinger H, Henze G. [Severe renal impairment of methotrexate elimination after high dose therapy]. KlinPadiatr 1994;206:319-26.
- Maiche AG, Lappalainen K, Teerenhovi L. Renal insufficiency in patients treated with high dose methotrexate. ActaOncol 1988;27:73-4.
- Mashhadi MA, Kaykhaei MA, Sanadgol H. Low Prevalence of High-dose Methotrexate Nephropathy in Patients With Malignancy. IJKD 2012;6:106-9.
- Nateghian A, Robinson JL, Arjmandi K, Vosough P, Karimi A, Behzad A, et al. Epidemiology of vancomycin-resistant enterococci in children with acute lymphoblastic leukemia at two referral centers in Tehran, Iran: a descriptive study. Int J Infect Dis 2011;15:e332–e335.
- Sayehmiri K, Alimoghaddam K, Ghavamzadeh A. Comparison of Prognostic Factors and Death Hazard Function of Acute Myeloid Leukemia (AML) and Acute Lymphoblastic Leukemia (ALL) Pateints after Bone Marrow Transplantation. DOAJ 2010;4(2):19-28.
- Sayehmiri K, Eshraghian MR, Mohammad K, Alimoghaddam K, RahimiForoushani A, Zeraati H, et al. Prognostic factors of survival time after hematopoietic stem cell transplant in acute lymphoblastic leukemia patients: Cox proportional hazard versus accelerated failure time models. J ExpClin Cancer Res 2008;27:74.
- Silverman LB, Gelber RD, Dalton VK, Asselin BL, Barr RD, Clavell LA, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood.2001;97(5):1211–8.
- Gleeson HK, Shalet SM. Endocrine complications of neoplastic diseases in children and adolescents. CurrOpinPediatr2001;13(4):346–51.
- Trimis G, Moschovi M, Papassotiriou I, Chrousos G, Tzortzatou-Stathopoulou F. Early indicators of dysmetabolic syndrome in young survivors of acute lymphoblastic leukemia in childhood as a target for preventing disease.J PediatrHematolOncol2007;29(5):309–14.
- Reisi N, Azhir A, Hashemipour M, Raeissi P, Amini A, Moafi A. The metabolic syndrome in survivors of childhood acute lymphoblastic leukemia in Isfahan, Iran. J Res Med Sci 2009;14(2):111-16.
- Meyer B, Hagen W, Scheithauer W, Ohler L, KornekGV. L-Asparaginase-associated hyperlipidemia withhyperviscosity syndrome in a patient with T-celllymphoblastic lymphoma. Annals of Oncology 2003;14(4): 658-9.
- Athale UH, Chan AK. Thrombosis in children withacute lymphoblastic leukemia Part III. Pathogenesis ofthrombosis in children with acute lymphoblasticleukemia: effects of host environment. Thromb Res2003;111(6):321-7.
- Cremer P, Lakomek M, Beck W, Prindull G. The effectof L-ASP on lipid metabolism during inductionchemotherapy of childhood ALL. European journal ofpediatrics 1998;147(1):64-7.
- Arzanian MT, Eghbali A, Alavi S, Shamsian BSH, Malek F,Azargashb E. L-Asparginase effect with 6000U/m2 on lipid profile in children with acute lymphoblastic leukemia. SJIBTO 2009;6(2):85-93.
- Mousavi SM, Pourfeizi A, Dastgiri S. Childhood cancer in Iran. J PediatrHematolOncol 2010;32:376–382.
- Chessells JM. Relapsed lymphoblastic leukaemia in children: a continuing challenge. Br J Haematol 1998;102:423–438.
- Gaynon PS. Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol 2005;131:579–587.
- Tissing WJ, Meijerink JP, den Boer ML, Pieters R. Molecular determinants of glucocorticoid sensitivity and resistance in acute lymphoblastic leukemia.Leukemia 2003;17:17–25.
- van Rossum EF, Koper JW, van den Beld AW, Uitterlinden AG, Arp P, Ester W, et al. Identification of theBclI polymorphism in the glucocorticoid receptor gene: association with sensitivity to glucocorticoids in vivo and body mass index. ClinlEndocrinol 2003;59:585–592.
- Weaver JU, Hitman GA, Kopelman PG. An association between a Bc1I restriction fragment length polymorphism of the glucocorticoid receptor locus and hyperinsulinaemia in obese women. J MolEndocrinol 1992;9:295–300.
- Buemann B, Vohl MC, Chagnon M, Chagnon YC, Gagnon J, Pérusse L, et al.Abdominal visceral fat is associated with a BclI restriction fragment length polymorphism at the glucocorticoid receptor gene locus. Obes Res 1997;5:186–192.
- Koper JW, Stolk RP, de Langr P, Huizenga NA, Molijn GJ, Pols HA, et al. Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet 1997;99:663–668.
- Huizenga NA, Koper JW, De Lange P, Pols HA, Stolk RP, Burger H, et al. A polymorphism in the glucocorticoid receptor gene may be associated with an increased sensitivity to glucocorticoids in vivo. J ClinEndocrinolMetab 1998;83:144–151.
- van Rossum EF, Koper JW, Huizenga NA, Uitterlinden AG, Janssen JA, Brinkmann AO, et al. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes 2002;51:3128–3134.
- Namazi S, Zareifar S, Monabati A, Ansari Sh, Karimzadeh I. Evaluating the Effect of 3 Glucocorticoid Receptor Gene Polymorphisms on Risk of Relapse in 100 Iranian Children
With Acute Lymphoblastic Leukemia: A Case-Control Study. ClinTher 2011;33(3):280-90.
ورود / ثبت نام