سندرم MONA
Multicentric Osteolysis, Nodulosis, and Arthropathy Syndrome

شاهین اسعدی (دانشجوی دکتری تخصصی ژنتیک پزشکی)
کلیاتی از سندرم MONA
سندرم MONA یک بیماری نادر ارثی ژنتیکی است که با کاهش بافت استخوانی (اُستئولیز) بهویژه در دستها و پاها مشخص میشود.
علائم و نشانههای بالینی سندرم MONA
سندرم MONA در اغلب موارد با از دست دادن بافت استخوان در دست و پا شروع میشود که منجر به حس درد و محدودیت حرکتی میگردد. ناهنجاریهای استخوانی نیز در این سندرم بعداً میتواند به سایر نقاط بدن گسترش یابد و با مشکلات مفصلی (آرتروپاتی) در آرنج، شانه، زانو، باسن و ستون فقرات همراه باشد.
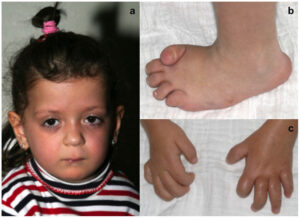
شکل 1: تصویر کودک مبتلا به سندرم MONA همراه با اختلالات مربوطه در دستها و پاها
در اکثر افراد مبتلا به سندرم MONA، تراکم کم مواد معدنی استخوان (اُستئوپنی) و پوکی استخوان در سراسر اسکلت بدن ایجاد میشود. این ناهنجاریها، استخوانها را شکننده و شکنندهتر میکند و میتواند منجر به کوتاهی قد و قامت مبتلایان شود. علاوه بر این، در بسیاری از افراد مبتلا به سندرم MONA گرههای زیرجلدی که نودولهای غیر قابل تشخیص زیرپوستی هستند بهویژه در کف پاها ایجاد میشود. برخی از افراد مبتلا به سندرم MONA دارای اختلالات پوستی مانند پوست تیره، ضخیم و چرب هستند. سایر ویژگیهای سندرم MONA میتواند شامل کدورت قرنیه چشم، رشد بیش از حد مو (هایپرتریچوزیس)، رشد بیش از حد لثهها، اختلالات قلبی و ویژگیهای مشخص صورت (چهره خشن) باشند.
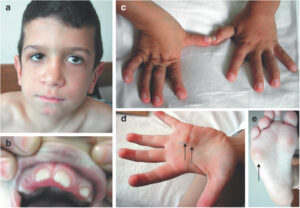
شکل 2: تصویری از اختلالات کودک مبتلا به سندرم MONA
علتشناسی سندرم MONA
سندرم MONA در اثر جهش ژن MMP2 که در بازوی بلند کروموزوم شماره 16 بهصورت 16q12.2 مستقر است، ایجاد میشود. این ژن دستورالعملهای لازم برای سنتز آنزیمی به نام ماتریکس متالوپپتیداز 2 را فراهم میکند که عملکرد اولیه این آنزیم به عنوان پروتئین کلاژن نوع IV است. کلاژن نوع IV جزء اصلی غشاء ساختاری زیرین سلول است که بسیاری از بافتهای بدن را پشتیبانی میکند. فعالیت ماتریکس متالوپپتیداز 2 برای توابع مختلف بدن از جمله بازسازی استخوان حائز اهمیت است که یک فرآیند طبیعی است و طی آن، استخوان قدیمی یا کهنه تجزیه و استخوان جدید یا جوان برای جایگزینی، ایجاد میشود.
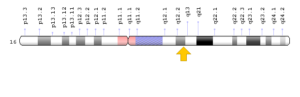
شکل 3: نمای شماتیک از کروموزوم شماره 16 که ژن MMP2 در بازوی بلند این کروموزوم بهصورت 16q12.2 مستقر است
جهشهای ژن MMP2 که سندرم MONA را ایجاد میکنند، از فعالیت آنزیم ماتریکس متالوپپتیداز 2 بهطور کامل جلوگیری میکنند و عملکرد طبیعی پروتئین کلاژن نوع IV را مختل میسازند. هنوز مشخص نیست که از دست دادن فعالیت آنزیم ماتریکس متالوپپتیداز 2 چگونه منجر به ویژگیهای خاص سندرم MONA میشود. محققان معتقدند که جهش در ژن MMP2 به نحوی توازن سنتز استخوان جدید و تجزیه استخوانهای کهنه و قدیمی را در طی فرآیند بازسازی استخوان مختل میکند که همین امر منجر به از دست دادن بافتهای استخوانی میشود. با این حال، چگونگی کمبود آنزیم ماتریکس متالوپپتیداز 2 در ایجاد گرههای زیرجلدی و ناهنجاریهای پوستی در سندرم MONA هنوز ناشناخته است.
سندرم MONA از الگوی توارثی اتوزومال مغلوب پیروی میکند؛ بنابراین برای ایجاد سندرم MONA دو نسخه از ژن جهشیافته MMP2 (یکی از پدر و دیگری از مادر) موردنیاز است و شانس داشتن فرزند مبتلا به سندرم MONA در این حالت، برای هر بارداری احتمالی به میزان 25% است.
فراوانی سندرم MONA
سندرم MONA یک اختلال ژنتیکی نادر است که فرکانس شیوع آن در جهان مشخص نیست. این سندرم در چندین جمعیت از سراسر جهان در ادبیات پزشکی گزارش شده است.
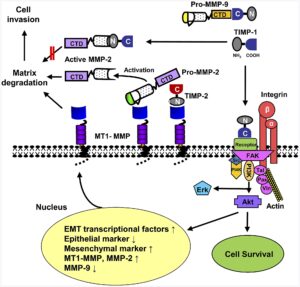
شکل 4: شماتیکی از مسیر مولکولی ژن MMP2
تشخیص سندرم MONA
سندرم MONA بر اساس یافتههای بالینی و فیزیکی مبتلایان و برخی آزمایشهای پاتولوژیکی تشخیص داده میشود. بهکارگیری تکنیکهای تصویربرداری رادیولوژیکی مانند پراش اشعه ایکس، اسکن ایزوتوپ استخوان و سونوگرافی نیز در تشخیص اختلالات اسکلتی سندرم MONA مفید هستند. قطعیترین روش تشخیص سندرم MONA، آزمایش ژنتیک مولکولی برای ژن MMP2 بهمنظور بررسی وجود جهشهای احتمالی است. تشخیص پیش از تولد نیز با استفاده از مایع آمنیوسنتز و یا نمونهبرداری از پرزهای کوریونی جفت جنین امکانپذیر است.
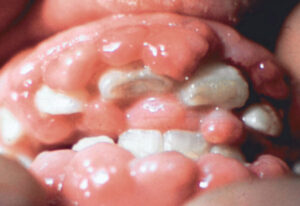
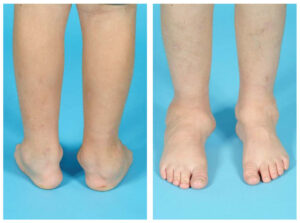
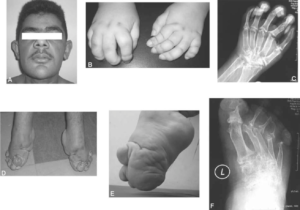
شکل 5: نمای کلی از اختلالات مربوط به سندرم MONA
مسیرهای درمانی سندرم MONA
استراتژی درمان و مدیریت سندرم MONA بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص اطفال، متخصص ارتوپدی، متخصص تغذیه، متخصص پوست و مو، متخصص قلب، متخصص چشم و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان قاطعی برای سندرم MONA وجود ندارد و تمامی اقدامات بالینی بهمنظور تخفیف رنج مبتلایان است. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند، از جایگاه ویژهای برخوردار است.
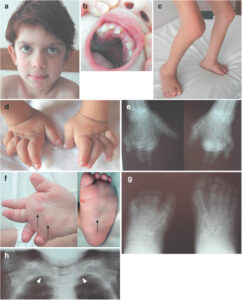
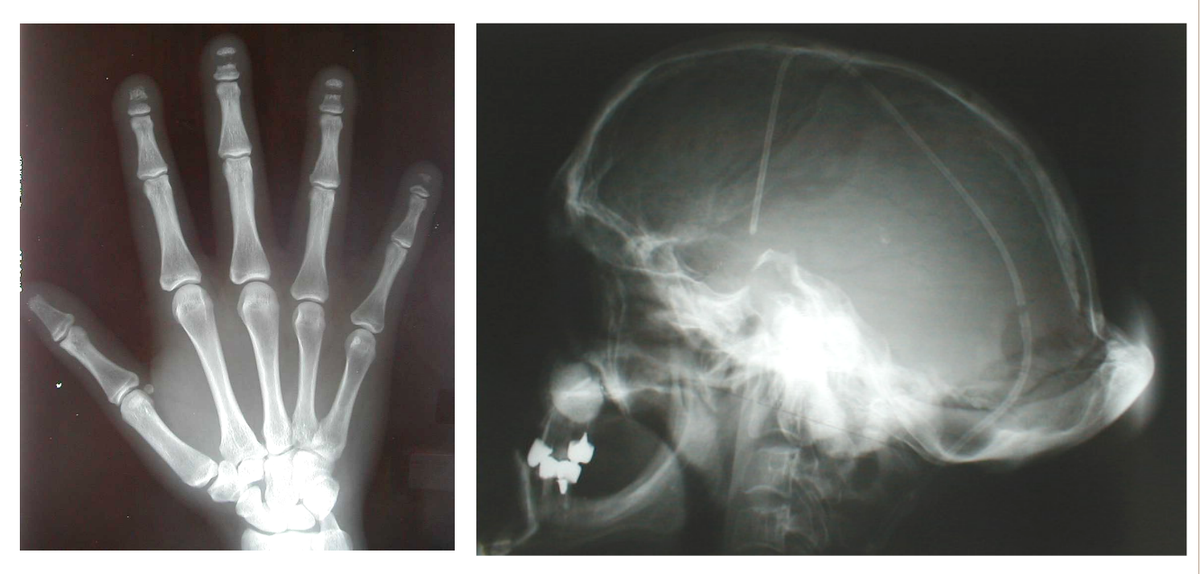
شکل 6: تصاویر رادیولوژی از اختلالات کودک مبتلا به سندرم MONA
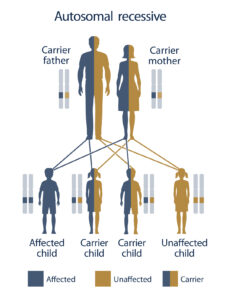
شکل 7: نمای شماتیک از الگوی تواثی اتوزومال مغلوب که سندرم MONA از این الگو پیروی میکند
منبع:
دکتر اسعدی شاهین، دکتر ملتیار حسن، دکتر روشنروان ندا، دکتر داداشپور مهدی، جمالی مهسا، محمدزاده حمیده، فتاحی محیا، کتاب پاتولوژی در ژنتیک پزشکی جلد 6 (M-Y)، صفحات 546-537، انتشارات کتب دانشگاهی عمیدی، بهار 1397.
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام