سندرم کوتیس لاکسا
Cutis Laxa Syndrome

شاهین اسعدی (کارشناس ارشد ژنتیک)
کلیاتی از سندرم کوتیس لاکسا
سندرم کوتیس لاکسا یک اختلال ژنتیکی در بافت همبند است که چارچوب حمایتی بدن را تشکیل میدهد. بافت همبند، ساختار و قدرت را به عضلات، مفاصل، اندامها و پوست میرساند.
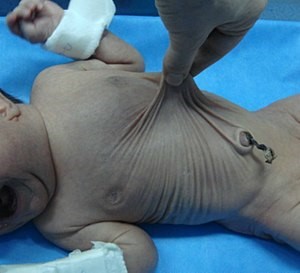
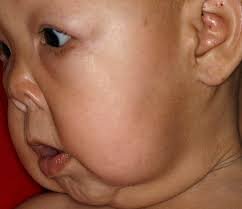
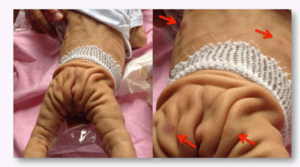
شکل 1: تصاویری از نوزادان مبتلا به سندرم کوتیس لاکسا همراه با اختلال پوستی
علائم و نشانههای بالینی سندرم کوتیس لاکسا
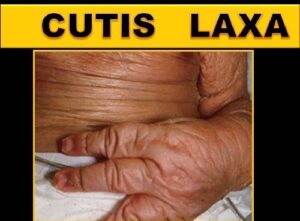
اصطلاح “cutis laxa” به زبان لاتین به پوست شلشده و یا ضعیف اشاره میکند و این شرایط با پوستی که چروکیده و غیرالاستیک است، مشخص میشود. پوست بسیار چروکیده ممکن است بر روی گردن و در زیر بغل و کشاله ران ظاهر شود.
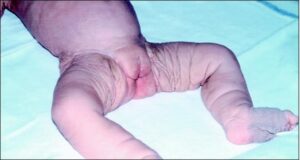
Cutis laxa همچنین میتواند بافت همبند در سایر قسمتهای بدن، از جمله قلب، رگهای خونی، مفاصل، روده و ریهها را تحت تأثیر قرار دهد. این اختلال میتواند مشکلات قلبی و باریک شدن غیرطبیعی، انحراف و یا پاره شدن عروق خونی قلب را ایجاد کند. افرادی که تحت تأثیر قرار گرفتهاند ممکن است پوسته نرم در شکم، دور کمر و یا در اطراف فتق ناف داشته باشند. کیسههایی که دیورتیکولا نامیده میشوند نیز میتوانند در دیوارههای اندامهای خاص مانند مثانه و روده ایجاد شوند. در دوران کودکی، در برخی از افراد مبتلا به این سندرم، یک بیماری ریوی به نام اِمفیزما ایجاد میشود که تنفس را دشوار میکند. علائم و نشانههای cutis laxa بسته به اینکه کدام اندامها و بافتها تحت تأثیر قرار میگیرند، میتوانند از خفیف تا تهدیدکننده زندگی متغیر باشند.
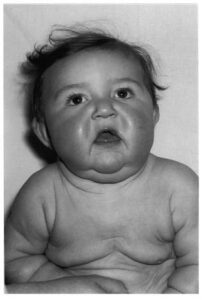
شکل 2: تصاویری از کودکان مبتلا به سندرم کوتیس لاکسا همراه با اختلالات مربوطه در پوست
محققان چندین شکل مختلف از کوتیس لاکسا را شناسایی کردهاند؛ این اَشکال اغلب توسط الگوی ارثی خود متمایز میشوند که شامل اتوزومی مغلوب و اتوزوم غالب یا وابسته به ایکس میشود. بهطور کلی، فرم اتوزومال مغلوب از سندرم کوتیس لاکسا، شدت بیشتری نسبت به فرم اتوزومال غالب از این سندرم دارد. علاوه بر ویژگیهایی که در بالا شرح داده شد، برخی از افراد مبتلا به سندرم کوتیس لاکسا با فرم اتوزومال مغلوب علائمی مانند تأخیر تکاملی، اختلالات ذهنی، تشنج و مشکلات حرکتی که میتواند در طول زمان بدتر شود را نیز تجربه میکنند.
فرم وابسته به ایکس سندرم کوتیس لاکسا اغلب با نام سندرم اکسیپیتال هورن شناخته میشود. این اختلال یک نوع خفیف از سندرم مِنکز است که بر سطوح مِس در بدن تأثیر میگذارد. سندرم اکسیپیتال هورن علاوه بر سفتی و غیراِلاستیکی شدن پوست با انباشت کلسیم در استخوان پایه جمجمه (استخوان اکسیپیتال), موی زِبر و شُلی مفاصل همراه است.
شکل 3: نمایی دیگر از اختلال پوستی در نوزاد مبتلا به سندرم کوتیس لاکسا
علتشناسی
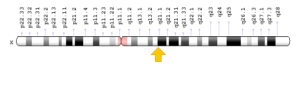
سندرم کوتیس لاکسا در اثر جهش ژن ATP6V0A2 که در بازوی بلند کروموزوم شماره 12 بهصورت 12q24.31 مستقر است، ژن ATP7A که در بازوی بلند کروموزوم جنسی X بهصورت Xq21.1 مستقر است، ژن EFEMP2 که در بازوی بلند کروموزوم شماره 11 بهصورت 11q13.1 مستقر است، ژن ELN که در بازوی بلند کروموزوم شماره 7 بهصورت 7q11.23 مستقر است و ژن FBLN5 که در بازوی بلند کروموزوم شماره 14 بهصورت 14q32.12 مستقر است، ایجاد میشود. بسیاری از این ژنها در تشکیل و عملکرد فیبرهای اِلاستیک نقش دارند که بستههای باریک پروتئینی هستند که قدرت و انعطافپذیری را برای بافت همبند در سراسر بدن فراهم میکنند. فیبرهای اِلاستیک به ارتجاع پوست، انبساط و انقباض ریهها و جریان خون در عروقی که با فشار بالا خون را به سایر نقاط بدن منتقل میکنند، اجازه میدهند.

شکل 4: نمای شماتیک از کروموزوم شماره 12 که ژن ATP6V0A2 در بازوی بلند این کروموزوم بهصورت 12q24.31 مستقر است
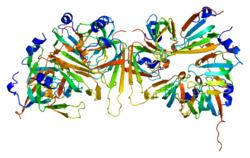
شکل 5: شماتیکی از ساختار بسته پروتئین ATP6V0A2
جزء اصلی فیبرهای اِلاستیک یک پروتئین به نام اِلاستین است که از ژن ELN تولید میشود. سایر پروتئینهایی که بهنظر میرسد نقش حیاتی در جمعآوری فیبرهای الاستـــــــیک دارند از ژنهای FBLN5 ,EFEMP2 ,ATP6V0A2 تولید میشوند. جهش در هر یک از این ژنها تشکیل، مونتاژ یا عملکرد فیبرهای اِلاستیک را مختل میکند. کمبود این فیبرها باعث تضعیف بافت همبند در پوست، عروق خونی، ریهها و سایر ارگانها میشود. این نقایص در بافت همبند اساس ویژگیهای اصلی سندرم کوتیس لاکسا هستند.
شکل 6: نمای شماتیک از کروموزوم جنسی X که ژن ATP7A در بازوی بلند این کروموزوم بهصورت Xq21.1 مستقر است

شکل 7: شماتیکی از ساختار بسته پروتئین ATP7A
سندرم اُکسیپیتال هورن در اثر جهش ژن ATP7A ایجاد میشود. این ژن دستورالعمل لازم برای سنتز پروتئینی را فراهم میکند که برای تنظیم مقادیر مس در بدن مهم است. جهش در ژن ATP7A منجر به انتشار ضعیف مس در سلولهای بدن میشود. کاهش انتشار مس میتواند فعالیتهای متعدد آنزیمهای حاوی مس را که برای ساختار و عملکرد استخوان، مو، پوست، عروق خونی و سیستم عصبی ضروری است، کاهش دهد. علائم و نشانههای سندرم اُکسیپیتال هورن ناشی از کاهش فعالیت این آنزیمهای حاوی مس است.

شکل 8: نمای شماتیک از کروموزوم شماره 11 که ژن EFEMP2 در بازوی بلند این کروموزوم بهصورت 11q13.1 مستقر است
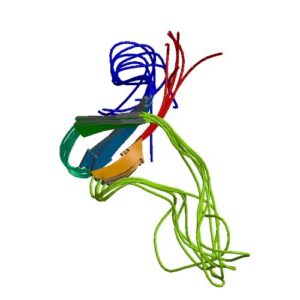
شکل 9: شماتیکی از ساختار بسته پروتئین EFEMP2
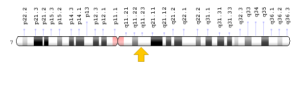
شکل 10: نمای شماتیک از کروموزوم شماره 7 که ژن ELN در بازوی بلند این کروموزوم بهصورت 7q11.23 مستقر است
شایان ذکر است جهش در ژنهایی که در بالا توضیح داده شده، تنها درصد کمی از موارد cutis laxa را تشکیل میدهند. جهش در ژنهای دیگر که بعضی از آنها هنوز شناسایی نشدهاند نیز میتوانند این وضعیت را ایجاد کنند.

شکل 11: نمای شماتیک از کروموزوم شماره 14 که ژن FBLN5 در بازوی بلند این کروموزوم بهصورت 14q32.12 مستقر است
سندرم کوتیس لاکسا با جهش در ژن ELN از الگوی توارثی اتوزومال غالب پیروی میکند، بنابراین برای ایجاد این سندرم یک نسخه از ژن جهشیافته ELN (اعم از پدر یا مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به این سندرم در حالت اتوزومی غالب، برای هر بارداری احتمالی به میزان 50% است. در موارد نادر نیز سندرم کوتیس لاکسا با جهش در ژن FBLN5 از الگوی توارثی اتوزومال غالب پیروی میکند.

شکل 12: نمای شماتیک از الگوی توارثی اتوزومال غالب که سندرم کوتیس لاکسا با جهش در ژنهای ELN و در برخی موارد ژن FBLN5 از این الگو تبعیت میکند
سندرم کوتیس لاکسا با جهش در ژنهای ATP6V0A2 و EFEMP2 از الگوی توارثی اتوزومال مغلوب پیروی میکند، بنابراین برای ایجاد این سندرم، دو نسخه از ژنهای جهشیافته ATP6V0A2 و EFEMP2 (یکی از پدر و دیگری از مادر) موردنیاز است و شانس داشتن فرزندی مبتلا به این سندرم در حالت اتوزومی مغلوب، برای هر بارداری احتمالی به میزان 25% میباشد.
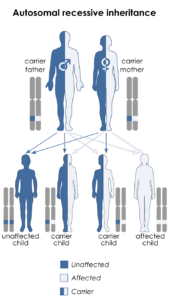
شکل 13: نمای شماتیک از الگوی توارثی اتوزومال مغلوب که سندرم کوتیس لاکسا با جهش در ژنهای ATP6V0A2 و EFEMP2 از این الگو تبعیت میکند
سندرم اکسیپیتال هورن با جهش در ژن ATP7A از الگوی توارثی وابسته به ایکس مغلوب پیروی میکند. این ژن در کروموزوم جنسی X واقع شده است. کروموزوم X یکی از دو کروموزوم جنسی است. در مردان (که فقط یک کروموزوم X دارند)، یک کپی تغییریافته از ژن در هر سلول برای ایجاد این وضعیت کافی است. در زنان (که دو کروموزوم X دارند)، جهش در هر دو نسخه ژن برای ایجاد اختلال کافی است. از آنجا که بعید است که زنان دو کپی تغییریافته از این ژن را داشته باشند، به همین دلیل زنان بهعنوان ناقل بیماری شناخته میشوند و مردان بیشتر از زنان تحت تأثیر اختلالات توارثی وابسته به ایکس قرار میگیرند. خصوصیات ارثی مرتبط با X این است که پدران نمیتوانند صفات مرتبط با X را به فرزندان پسر خود منتقل کنند.

شکل 14: نمای شماتیک از الگوی توارثی وابسته به ایکس مغلوب، مادر ناقل (سمت چپ) و پدر بیمار (سمت راست) که سندرم کوتیس لاکسا با جهش در ژن ATP7A از این الگو تبعیت میکند
فراوانی سندرم کوتیس لاکسا
سندرم کوتیس لاکسا اختلال ژنتیکی نادری است که تاکنون حدود 200 مورد مبتلا به آن از سراسر جهان در متون پزشکی گزارش شده است.
تشخیص
سندرم کوتیس لاکسا بر اساس یافتههای بالینی و کلینیکی مبتلایان و برخی آزمایشهای پاتولوژیکی تشخیص داده میشود. دقیقترین روش تشخیص این سندرم، آزمایش ژنتیک مولکولی برای ژنهای ATP6V0A2 ,ELN ,FBLN5 ,EFEMP2 ,ATP7A بهمنظور بررسی وجود جهشهای احتمالی میباشد.
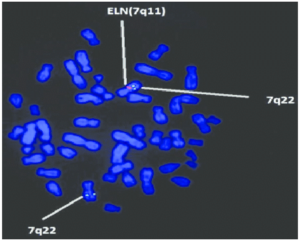
شکل 15: تصویر میکروسکوپیک از محل استقرار ژن ELN در بازوی بلند کروموزوم شماره 7 در منطقه q11.23 توسط تکنیک هیبریداسیون فلورسانس درجا (FISH)
مسیرهای درمانی
استراتژی درمان و مدیریت سندرم کوتیس لاکسا بهصورت علامتی و حمایتی است. درمان ممکن است با تلاش و هماهنگی تیمی از متخصصان منجمله متخصص پوست و مو، متخصص ارتوپدی، متخصص مغز و اعصاب، متخصص روده و گوارش، متخصص دستگاه ریوی و سایر متخصصان مراقبتهای بهداشتی انجام پذیرد. درمان استانداردی برای این سندرم وجود ندارد و تمامی اقدامات بالینی بهمنظور کاهش رنج مبتلایان میباشد. مشاوره ژنتیک نیز برای تمامی والدینی که طالب فرزندی سالم هستند، ضرورت دارد.

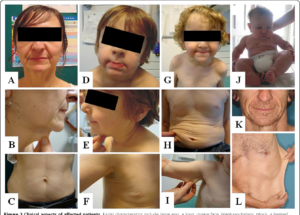
شکل 16: نمای کلی از اختلالات مربوطه در سندرم کوتیس لاکسا

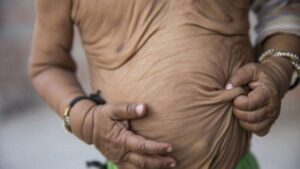
شکل 17: تصاویری از اختلال پوستی در مبتلایان سندرم کوتیس لاکسا
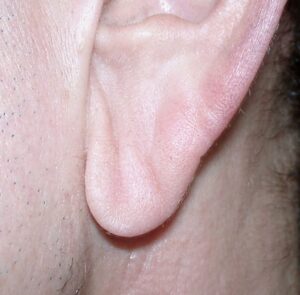
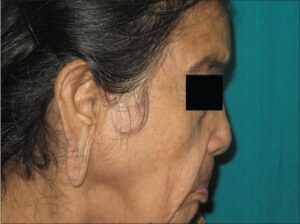
شکل 18: تصاویری از اختلال بافت همبند اضافی در گوشهای زن مبتلا به سندرم کوتیس لاکسا
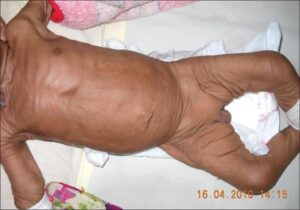
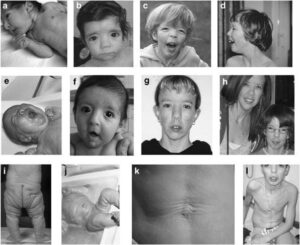
شکل 19: تصاویری از تمامی اختلالات مربوط به سندرم کوتیس لاکسا
منبع
اسعدی شاهین، قلیزاده شیوا، جمالی مهسا، علیپور شهریار، جوادی لیلا، صراطی نوری حامد، مهمان روش آیسان، سادهدل سمانه، کتاب پاتولوژی در ژنتیک پزشکی 11 (A-H), فصل سوم، صفحات 290-273، انتشارات کتب دانشگاهی عمیدی، 1397.
سندرم کمخونی دیاموند- بلکفان Diamond-Blackfan Anemia Syndrome
سندرم بال پروانهای Epidermolysis Bullosa (EB) Syndrome
https://rarediseases.org/rare-diseases/cutis-laxa/
برای دانلود پی دی اف برروی لینک زیر کلیک کنید
ورود / ثبت نام