سندرم کرایگلر نجار
Crigler-Najjar syndrome


شاهین اسعدی (دانشجوی ژنتیک مولکولی)، مهسا جمالی (کارشناس ارشد ژنتیک)
سندرم کرایگلر نجار یک اختلال ژنتیکی نادر است که نقص در کونژوگه کردن بیلیروبین در کبد با عدم توانایی تبدیل صحیح و دفع بیلیروبین از بدن همراه است، به عبارتی این سندرم در متابولیسم بیلیروبین تأثیر گذاشته و منجر به تجمع بیلیروبین با سطوح بالا در بدن میشود که همین امر باعث زردی غیرهمولیتیک در افراد مبتلا میشود. بیلیروبین که رنگدانه صفراوی نارنجی و زرد است، بهطور عمده محصول جانبی از فروپاشی طبیعی (دژنراسیون) سلولهای قرمز خون قدیمی و یا فرسوده (همولیز) میباشد.
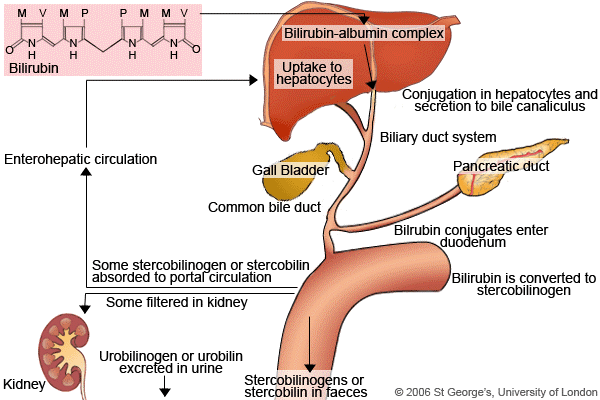
شکل 1: نمای شماتیک از کمپلکس بیلیروبین با آلبومین و نحوه عملکرد آن در بدن انسان
هموگلوبین موجود در گلبولهای قرمز پس از تخریب به یک مولکول هم و اسید آمینه تجزیه میشود. حلقه هم نیز تجزیه شده و تبدیل به یک مولکول بیلیوردین و نهایتاً تبدیل به بیلیروبین میشود. این بیلیروبین، بیلیروبین غیرکونژوگه بوده و در آب نامحلول است، لذا در ادرار و صفرا دفع نمیشود ولی محلول در چربی بوده و از سد خونی- مغزی و جفت عبور میکند (آسیب به مغز و جنین).
بیلیروبین غیرکونژوگه در داخل کبد توسط آنزیمUDP گلوکورونیل ترانسفراز با ۲ مولکول اسید گلوکورونیک پیوند برقرار کرده و تبدیل به بیلیروبین کونژوگه میشود. بیلیروبین کونژوگه محلول در آب میباشد. بیلیروبین بعد از کونژوگاسیون وارد صفرا و از آنجا وارد روده باریک شده و دفع میگردد. افراد مبتلا به سندرم کرایگلر نجار، توانایی تبدیل بیلیروبین غیرکونژوگه به بیلیروبین کونژوگه را ندارند، چراکه این افراد فاقد آنزیم کبدی خاص (UDP گلوکورونیل ترانسفراز) برای شکستن (سوختوساز) بیلیروبین هستند، بنابراین این افراد دارای سطوح بالایی از بیلیروبین غیرکونژوگه در جریان خون خواهند بود که منجر به زردی پوست و زردی کاسه چشم میگردد.
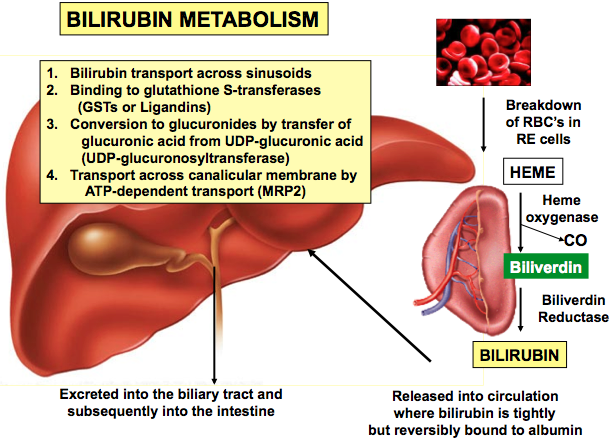
شکل 2: نمای شماتیک از متابولیسم بیلیروبین در کبد انسان
سندرم کرایگلر نجار به دو شکل میتواند انسان را درگیر کند؛ سندرم کرایگلر نجار نوع 1 و سندرم کرایگلر نجار نوع 2
سندرم کرایگلر نجار نوع 1
نوع ۱ با بالا بودن شدید بیلیروبین غیرکونژوگه در حد ۲۰ تا ۴۵ مشخص میشود. بیماری در دوره نوزادی ظاهر شده و تا پایان عمر باقی میماند. مشکل اصلی اینها فقدان آنزیم کبدی گلوکورونیل ترانسفراز است. اغلب بیلیروبین بالا باعث آسیب دائمی به دستگاه عصبی (اصطلاحاً بنام کرنایکتروس) میشود. زردی (jaundice) چند روز پس از تولد آغاز میشود. اگر پیوند کبد انجام نشود به علت ایجاد آنسفالوپاتی بیلیروبین دیررس که غالباً متعاقب یک بیماری تبدار روی میدهد، بیمار فوت میکند. اگر بیمار دچار آسیب عصبی نشده باشد انجام این اقدامات باعث بهبود وضعیت بیماران خواهد شد: نوردرمانی (نور آبی یا سفید ۱۲ ساعت در روز)، ترانسفوزیون خون، مهارکنندههای آنزیم همواکسیژناز مانند protoporphyrin،tin-mesoporphyrin، ترکیبات کلسیم خوراکی و پیوند کبد زودرس
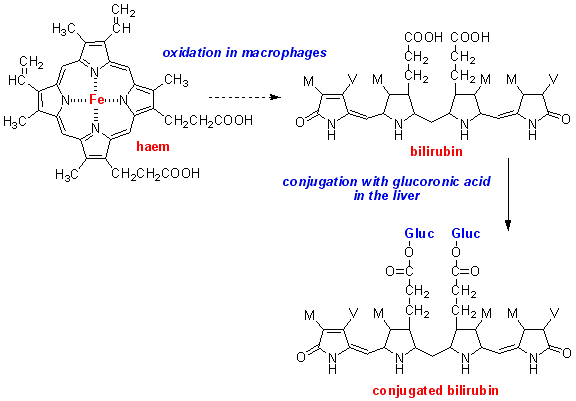
شکل 3: نمایی شماتیک از ساختار حلقوی بیلیروبین کونژوگه در کبد انسان
سندرم کرایگلر نجار نوع 2
در این بیماری نیز کمبود آنزیم گلوکورونیل ترانسفراز داریم (حدود ۱۰٪ انسان سالم) ولی شدت کمبود خفیفتر از نوع ۱ است، لذا بیلیروبین خیلی بالا نیست و مشکلات نورولوژیک (کرنایکتروس) در اینها شایع نیست. اگرچه معمولاً این بیماری را در سنین شیرخوارگی تشخیص میدهند اما گاهی تا سن جوانی هم تشخیص داده نمیشود. با تجویز فنوباربیتال (محرک آنزیم ترانسفراز)، میتوان سطح بیلیروبین سرم را به ۳ تا ۵ رساند. اگرچه بروز مشکلات عصبی در این نوع اندک است اما زردی نهتنها در شیرخوارگی، بلکه در بالغین، غالباً در شرایط بیماری همراه، گرسنگی یا به علت سایر عواملی که بهطور موقت زردی را افزایش میدهند، رخ داده است. درمان با فنوباربیتال بهطور گسترده بهکار میرود. یک دوز دارو هنگام خواب غالباً برای نگهداری غلظت بیلیروبین پلاسما در حد طبیعی کافی میباشد.
علائم و نشانههای بالینی سندرم کرایگلر نجار
علائم و نشانههای سندرم کرایگلر نجار نوع 1، اندکی پس از تولد ظاهر میشود. نوزادان مبتلا، زردی شدید و مداوم از پوست، غشاهای مخاطی و سفیدی چشم (یرقان) را آشکار میکنند. این علائم، پس از سه هفته اول زندگی باقی میمانند. نوزادان مبتلا به سندرم کرایگلر نجار نوع 1، در معرض خطر توسعه کرنایکتروس (کرنایکتروس یک بیماری عصبی است که بهطور بالقوه تهدیدکننده زندگی است و در آن سطوح سمی بیلیروبین در مغز تجمع مییابد و باعث آسیب به سیستم عصبی مرکزی میشود) و آنسفالوپاتی بیلیروبین در ماه اول زندگی هستند. نشانههای اولیه از کرنایکتروس ممکن است شامل فقدان انرژی (ضعف)، استفراغ، تب و تغذیه نامطلوب باشد، همچنین ممکن است این نوزادان علائمی مانند رفلکسهای مورو را نداشته باشند.

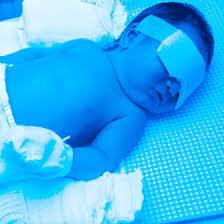
شکل 4: تصویر کودک مبتلا به سندرم کرایگلر نجار با میزان بیلیروبین بالا در خون که پوست زرد را نشان میدهد در مقابل کودک سالم
علاوه بر این، نوزادان مبتلا به سندرم کرایگلر نجار نوع 1، اسپاسم عضلانی خفیف تا شدید را تجربه خواهند کرد؛ از جمله اسپاسمی که در آن، سر و پاشنه پا خم شده و پشت بدن بهصورت قوس (کمانپیکری) است و یا حرکات غیرارادی کنترل نشده را تجربه میکنند.
شکل 5: نمای شماتیک از کودک با حالت مورو رفلکس
نوزادان مبتلا به سندرم کرایگلر نجار نوع 1 با دو حالت از کرنایکتروس مواجه میشوند، کرنایکتروس خفیف که منجر به اجرای نادرست مهارتهای حرکتی و کاهش رشد کامل مینای دندان میشود و کرنایکتروس شدید که منجر به از دست دادن حس شنوایی، مشکلات در ادراک حسی، تشنج آهسته و مداوم، نوشتن غیرارادی روی کاغذ در دوران مدرسه و حرکت پریشی از دست و پا یا کل بدن میشود. شایان ذکر است که موارد بسیار شدید از کرنایکتروس، میتواند تهدیدکننده حیات و زندگی باشد که منجر به مرگ انسان خواهد شد. اگرچه کرنایکتروس معمولاً در اوایل دوران نوزادی توسعه مییابد اما در برخی موارد افراد مبتلا به سندرم کرایگلر نجار نوع 1 ممکن است کرنایکتروس را تا اوایل دوران کودکی و حتی تا اوایل دوران بزرگسالی هم بروز ندهند.
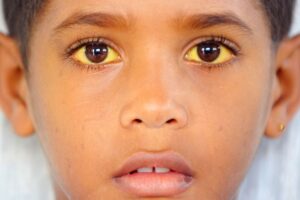
شکل 6: تصویر کودک مبتلا به سندرم کرایگلر نجار، به زردی کاسه چشمها دقت شود
سندرم کرایگلر نجار نوع 2 یک اختلال خفیفتر از نوع اول است که باعث زردی نوزادان میشود و این مکانیسم در طول زمان افزایش مییابد. این حالت بیشتر در زمانی رخ میدهد که کودک بیمار برای مدت طولانی گرسنگی داشته باشد و یا تحت بیهوشی عمومی بهمنظور جراحی عمومی ضروری پس از تولد قرار گیرد. زردی این نوزادان تا زمان بزرگسالی بهطور بارز مشهود نیست. کرنایکتروس در نوزادان مبتلا به سندرم کرایگلر نجار نوع 2، نادر است اما میتواند رخ دهد بهویژه موقعی که فرد مبتلا، برای مدت طولانی غذا نخورد و یا تحت بیهوشی عمومی قرار گیرد.
علت شناسی سندرم کرایگلر نجار
سندرم کرایگلر نجار توسط جهش در ژن UGT1A1 ایجاد میشود. ژن UGT1A1 در بازوی بلند کروموزوم شماره 2 بهصورت 2q37.1 مستقر است و شامل دستورالعمل خاصی برای سنتز آنزیم کبدی یوریدین به نام دی فسفات گلکورونوزیل ترانسفراز 1 میباشد که این آنزیم برای تبدیل بیلیروبین غیرکونژوگه به بیلیروبین کونژوگه و دفع آن از کبد موردنیاز است.
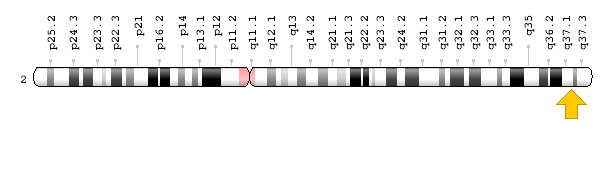
شکل 7: نمای شماتیک از محل استقرار ژن UGT1A1 در بازوی بلند کروموزوم شماره 2 بهصورت 2q37.1
فقدان جزئی یا کامل از این آنزیم منجر به تجمع بیلیروبین غیرکونژوگه در بدن میشود. بیلیروبین در گردش مایع پلاسمای خون به فرم کونژوگه میباشد که با پروتئین آلبومین ارتباط برقرار میکند و به فرم بیلیروبین غیرکونژوگه تبدیل میشود که نمیتواند در آب حل شود. بهطور معمول این بیلیروبین غیرکونژوگه توسط سلولهای کبدی گرفته میشود و با کمک آنزیم UGT1A1 تبدیل به فرم بیلیروبین گلوکورونید محلول در آب (بیلیروبین کونژوگه) میشود که از طریق صفرا در کیسه صفرا ذخیره شده و از قسمت بالایی روده کوچک (دئودنوم) وارد مدفوع شده و دفع میگردد، بنابراین جهش در ژن سنتزکننده آنزیم UGT1A1 منجر به عدم تبدیل بیلیروبین غیر کونژوگه به بیلیروبین کونژوگه میشود و در نتیجه بیلیروبین دفع نمیشود و در شریان خونی و کبد تجمع میکند و هنگامیکه سطح بیلیروبین بهاندازه زیاد افزایش پیدا کند، میتواند از سد خونی مغزی عبور کند و به بافت مغز و بافت عصبی نفوذ کند که منجر به بروز علائم عصبی مغزی در مبتلایان سندرم کرایگلر نجار خواهد شد. والدین کودکان مبتلا به سندرم کرایگلر نجار نوع 1، برخی از نقایص در سوختوساز بیلیروبین را از نظر بالینی و کلینیکی دارا هستند اما این والدین هیچ یافته فیزیکی از سندرم کرایگلر نجار را از خود نشان نمیدهند، چون آنها فقط یک کپی از ژن جهشیافته UGT1A1 (هتروزیگوت) را دارند. سندرم کرایگلر نجار از الگوی توارثی اتوزومال مغلوب پیروی میکند. اختلالات ژنتیکی مغلوب زمانی رخ میدهد که فرد، دو نسخه یا دو کپی از ژن تغییریافته را یکی از پدر و دیگری را از مادر به ارث ببرد. پدر و مادری که خویشاوند یکدیگر هستند، شانس بیشتری برای داشتن فرزندی با اختلالات ژنتیکی مغلوب خواهند داشت.
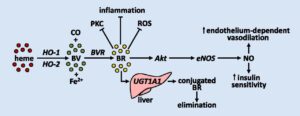
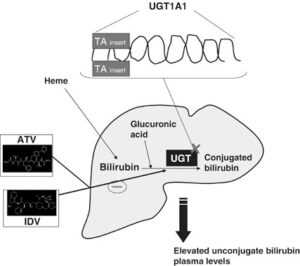
شکل 8: نمای شماتیک از مکانیسم عمل آنزیم UGT1A1 در سلولهای کبدی انسان
فراوانی شیوع سندرم کرایگلر نجار
سندرم کرایگلر نجار، مردان و زنان را به تعداد مساوی تحت تأثیر قرار میدهد. شیوع این سندرم حدوداً 1 در 750000 یا 1 در 1000000 تولد زنده انسان است.
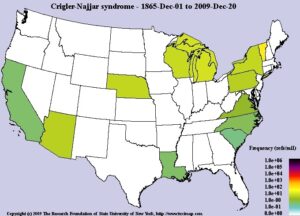
شکل 9: نمای شماتیک از فراوانی سندرم کرایگلر نجار در جهان از 1 دسامبر 1865 تا 20 دسامبر 2009
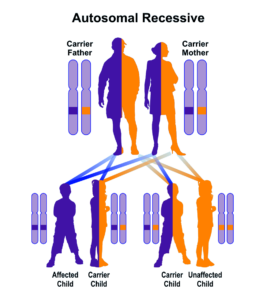
شکل 10: نمای شماتیک از الگوی توارثی اتوزومال مغلوب در سندرم کرایگلر نجار
تشخیص سندرم کرایگلر نجار
تشخیص این سندرم ممکن است در چند روز اول تولد نوزاد، با مشاهده زردی مداوم و مشکوک صورت پذیرد. همچنین تشخیص سندرم کرایگلر نجار میتواند بر اساس ارزیابی بالینی، یافتههای مشخص و آزمایشهای تخصصی انجام گیرد؛ بهعنوان مثال در نوزادان مبتلا به این اختلال، آزمایش خون میزان غیرطبیعی بیلیروبین غیرکونژوگه را نشان میدهد، بااینحال، آزمایش ژنتیک مولکولی قطعیترین تشخیص سندرم کرایگلر نجار را اعلام میکند. آزمایش ژنتیک مولکولی، میتواند جهش در ژن UGT1A1 را تشخیص دهد ولی این روش آزمایش قابل دسترس برای همگان نیست و فقط در مراکز تشخیص ژنتیکی قابل سرویسدهی است.
مسیرهای درمانی سندرم کرایگلر نجار
درمان اصلی این سندرم با هدف کاهش سطح بیلیروبین غیرکونژوگه در خون انجام میگیرد. شروع درمان زودهنگام برای سندرم کرایگلر نجار نوع 1 بهمنظور جلوگیری از توسعه کرنایکتروس در طول چند ماه اول زندگی ضروری است. ازآنجاکه سندرم کرایگلر نجار نوع 2 دارای علائم بالینی خفیفتری است، استفاده از داروی فنوباربیتال میتواند مؤثر باشد.
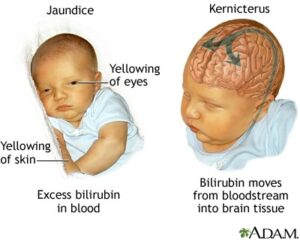
شکل 11: نمای شماتیک از کودک مبتلا به یرقان همراه با نفوذ بیلیروبین به بافت مغزی که منجر به اختلال کرنایکتروس میشود
درمان اصلی برای سندرم کرایگلر نجار نوع 1، فتوتراپی تهاجمی است. در طی این فرایند، پوست لخت در معرض شدید نور قرار میگیرد و همزمان از چشمها محافظت میشود. مسن بودن افراد مبتلا به سندرم کرایگلر نجار، پوست ضخیم و افزایش توده بدن باعث تأثیرپذیری کم فتوتراپی برای جلوگیری از کرنایکتروس میشود. برخی پزشکان استفاده از دیودهای ساطعکننده نور به رنگ آبی (LED) را برای کاهش سطح بیلیروبین خون توصیه میکنند، اما این تکنیک بهطور گسترده در جامعه پزشکی استفاده نمیشود.

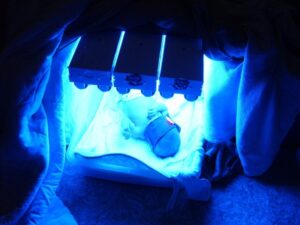
شکل 12: تصاویری از درمان نوزادان مبتلا به سندرم کرایگلر نجار با دیودهای ساطعکننده نور آبی (LED)
قرار گرفتن پوست در معرض نور خورشید برای کاهش سطح بیلیروبین خون بسیار مؤثر است. پیوند کبد تنها درمان قطعی برای افراد مبتلا به سندرم کرایگلر نجار نوع 1 میباشد، اما خود پیوند کبد دارای مشکلات فراوانی است از قبیل هزینه بالا، محدود بودن اهداکننده، نیاز به استفاده طولانی از داروهای سرکوبکننده سیستم ایمنی و یا رد پیوند. پزشکان معتقدند، پیوند کبد باید قبل از دوران نوجوانی انجام پذیرد تا از آسیب مغزی ناشی از کرنایکتروس پیشگیری شود. مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها توصیه میشود.
درمان تحقیقاتی سندرم کرایگلر نجار
ژندرمانی بهعنوان یک روش تحقیقاتی برای درمان افراد مبتلا به سندرم کرایگلر نجار میتواند مورد استفاده قرار گیرد. در ژندرمانی، ژن معیوب بیمار با ژن سالم جایگزین میشود که این نوع انتقال میتواند بهصورت دائمی باشد و منجر به درمان طولانیمدت بیماران شود.
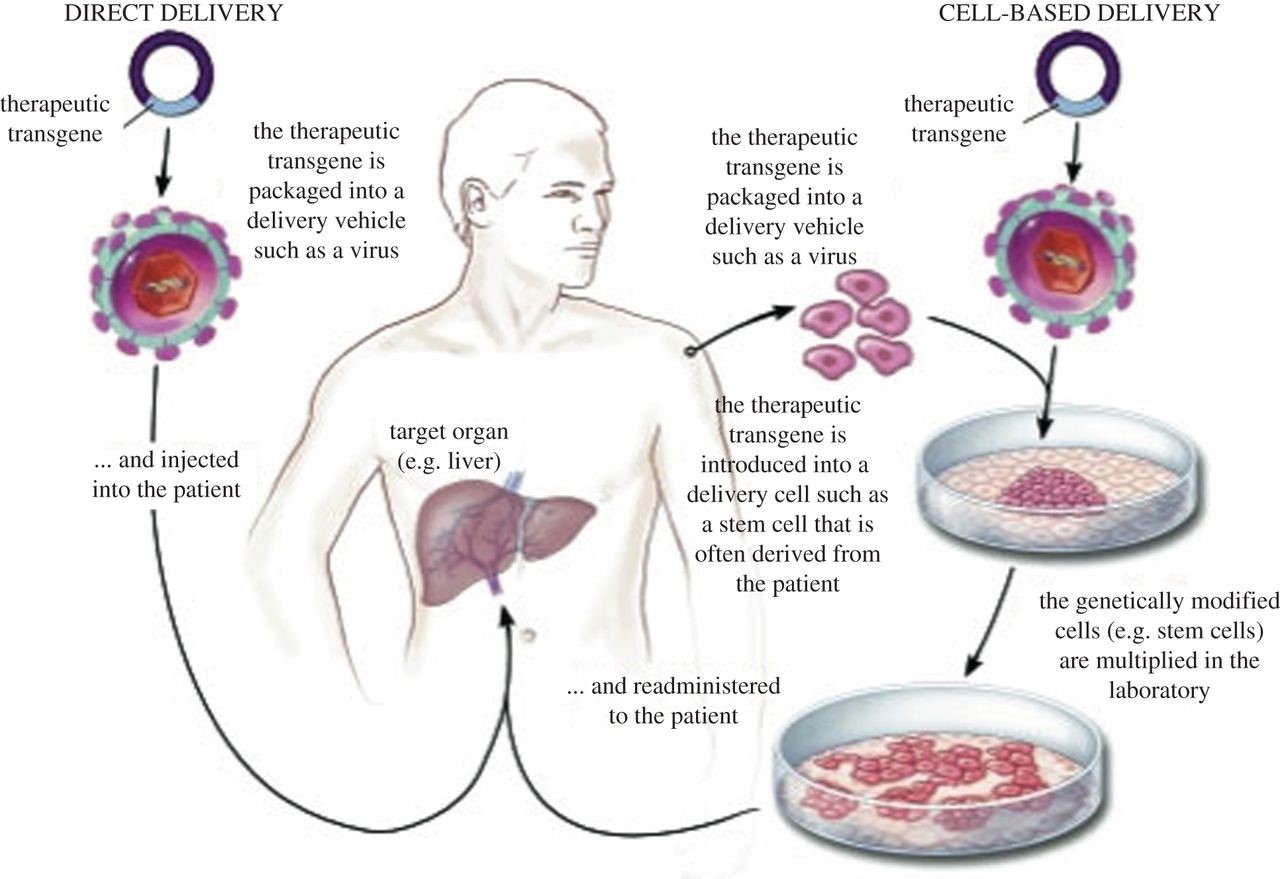
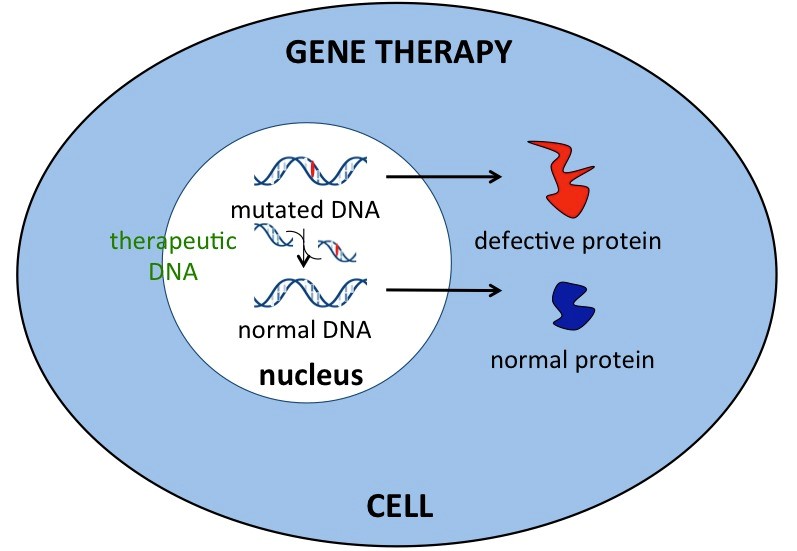
شکل 13: نمای شماتیک از ژندرمانی در انسان
تاریخچه سندرم کرایگلر نجار
سندرم کرایگلر نجار، اولین بار در شش نوزاد از سه زوج که همگی باهم خویشاوند بودند، شناسایی شد. این سندرم در سال 1952 توسط دکتر جان فلدینگ کرایگلر پزشک متخصص اطفال آمریکایی و دکتر ویکتور اسد نجار پزشک متخصص اطفال آمریکایی لبنانیتبار گزارش گردید. در سال 1962، دکتر آریاس یک نسخه خفیفتر از این سندرم را گزارش کرد که امروزه آن را سندرم کرایگلر نجار نوع 2 میشناسیم.

شکل 14: تصاویر دکتر جان فلدینگ کرایگلر و دکتر ویکتور اسد نجار، کاشفان سندرم کرایگلر نجار
References:
- Jansen PL (December 1999). “Diagnosis and management of Crigler–Najjar syndrome”. European journal of pediatrics. 158 (Suppl 2): S89–S94.
- Chowdhury، J. R.; Wolkoff، A. W.; Chowdhury، N. R.; Arias، I. M.: “Hereditary jaundice and disorders of bilirubin metabolism.” In: Scriver، C. R.; Beaudet، A. L.; Sly، W. S.; Valle، D. (eds.): The Metabolic and Molecular Bases of Inherited Disease. Vol. 2. New York: McGraw-Hill (8th ed.) 2001. Pp. 3063–3101.
- Fox IJ، Chowdhury JR، Kaufman SS، Goertzen TC، Chowdhury NR، Warkentin PI، Dorko K، Sauter BV، Strom SC (May 1998).
- Toietta G، Mane VP، Norona WS، Finegold MJ، Ng P، Mcdonagh AF، Beaudet AL، Lee B (March 2005).
- Crigler JF Jr، Najjar VA (February 1952). “Congenital familial nonhemolytic jaundice with kernicterus; a new clinical entity”. AMA American Journal of Diseases of Children. 83 (2): 259–60.
- Downs E، Gourley GR. Neonatal Jaundice and Disorders of Bilirubin Metabolism. In: Nathan and Oski’s Hematology of Infancy and Childhood، 7th ed. Orkin SH، Nathan DG، Ginsburg D، Look AL، Fisher DE، Lux SE، editors. 2015 Elsevier Saunders، Philadelphia، PA. pp.101-127.e12.
- Askari FK. Crigler-Najjar Syndrome. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia، PA. 2003:337.
- Behrman RE، Kliegman RM، Jenson HB. Eds. Nelson Textbook of Pediatrics. 17th ed. Elsevier Saunders. Philadelphia، PA; 2005:1320-1321.
- Scriver CR، Beaudet AL، Sly WS، et al. Eds. The Metabolic Molecular Basis of Inherited Disease. 8th ed. McGraw-Hill Companies. New York، NY; 2001:3078-3087.
- Memon N، Weinberger BI، Hegyi T، Aleksunes LM. Inherited disorders of bilirubin clearance. Pediatr Res. 2016;79(3):378-386.
- Van Dijk R، Beuers U، Bosma PJ. Gene replacement therapy for genetic hepatocellular jaundice. Clin Rev Allergy Immunol. 2015;48:243-253.
- Sticova E، Jirsa M. New insights in bilirubin metabolism and their clinical implications. World J Gastroenterol. 2013;196398-6407.
- Kadakol A، Ghosh SS، Sappal BS، et al. Genetic lesions of bilirubin uridine-diphospho- glucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes correlation of genotype to phenotype. Human Mutation. 2000;16:297-306.
- Jansen PL. Diagnosis and management of Crigler-Najjar syndrome. Eur J Pediatr. 1999;158 Suppl 2:S89-94.
- Arias IM، Gartner LM، Cohen M، et al. Chronic nonhemolytic unconjugated hyperbilirubinemia with glucuronosyltransferase deficiency. Am J Med. 1969;47:395-409.
- Crigler FJ، Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics. 1952;10:169-170.
بیلیروبین مستقیم بیشتر از بیلیروبین توتال؟
بررسی بیوسنتز و اختلالات ناشی از متابولیسم هم (Heme)
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام