سندرم کاودن
Cowden Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، زینب غلامی (دانشجوی هماتولوژی)، دکتر سید مجتبی محدث اردبیلی (متخصص ژنتیک پزشکی)
دانشگاه علوم پزشکی تبریز، مرکز تشخیص ژنتیک پزشکی و مشاوره ژنتیک دکتر مجتبی محدث اردبیلی


سندرم کاودن، به عنوان سندرم هامارتوم چندگانه شناخته شده است. این بیماری یک اختلال نادر ژنتیکی است که به صورت اتوزومال غالب به ارث میرسد. این سندرم با چند غده مانند تومور به نام هامارتومها و افزایش خطر ابتلا به انواع خاصی از سرطان همراه است. سندرم کاودن، با جهش در ژن PTEN که یک ژن سرکوبگر تومور است باعث عملکرد و سنتز نادرست پروتئین PTEN میشود که همین امر منجر به بیشفعالی در کودکان از مسیر MTOR خواهد شد. این جهش منجر به بروز ویژگیهای مشخصه از جمله ماکروسفالی، پولیپ روده، هامارتوماتوز، تومورهای خوشخیم متعدد پوست، پاپیلوماتوز، کراتوسیز و گانگلیوسیتومای دیسپلاستیک مخچه میشود.
علاوه بر اینها بیماران مبتلا ، مستعد ابتلا به سرطان پستان، کارسینوم فولیکولر تیروئید و سرطان آندومتر میباشند.
علائم و نشانها:
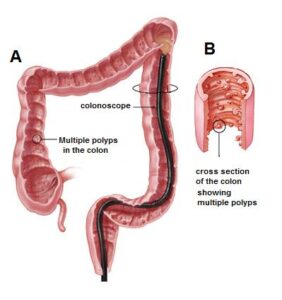
ویژگیهای بالینی سندرم کاودن متنوع هستند از جمله سرطان پستان، اندومتر، تیروئید، سرطان روده بزرگ، ویژگیهای پوستی مانند پاپیلوماز دهان و پوست، ویژگیهای دستگاه گوارش مانند پولیپ مخاط و از جمله هامارتوم، و ویژگیهای عصبی مانند بیماری Lhermitte-Duclos.
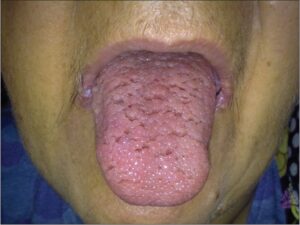
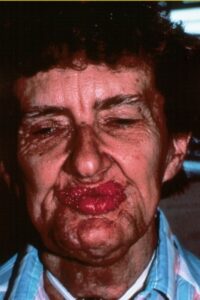
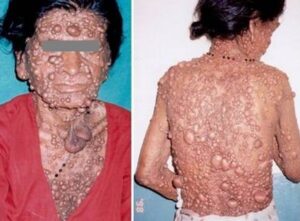
هامارتومهای کوچک مشخصه سندرم کاودن هستند. غدههای غیرسرطانی که معمولاً بیشتر پوست و غشاهای مخاطی مثل پوشش دهان و بینی را دربر دارند، میتوانند در دستگاه گوارش و دیگر قسمتهای بدن بوجود آیند. این غدهها تا حد زیادی خوشخیم هستند، با این حال افراد مبتلا به سندرم کاودن، همیشه مستعد خطر ابتلا به سرطان پستان، تیروئید و سرطان رحم میباشند.
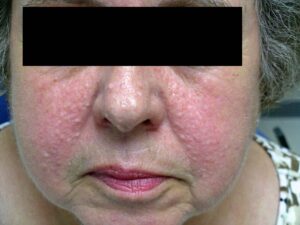
افراد مبتلا به سندرم کاودن تا 75 درصد، دارای شرایط خوشخیم غدههای پستان مانند، هیپرپلازی داکتال، پاپیلوماتوزیز، آدنوزیز، اینتراداکتال، آتروفی لوبولار، فیبروآدنومها و تغییرات فیبروکیستیک هستند. فقط 10 درصد از افراد مبتلا به سندرم کاودن مستعد ابتلا به سرطان تیروئید میباشند. علاوه بر این بیش از نیمی از کسانی که، تحت تأثیر سندرم کاودن قرار دارند، دارای آدنوم فولیکولار یا گواتر ندولار تیروئید هستند. بدخیمیهای دیگر که به نظر میرسد با سندرم کاودن همراهند عبارتند از سرطان آندومتر و سرطان کلیوی، و همچنین علائم و نشانههای دیگر سندرم کاودن شامل سر بزرگ، تومور مغزی غیرسرطانی نادر بنام بیماری Lhermitte-Duclos و همینطور آکانتوز گلیکوژنیک مری است. اکثر افراد مبتلا به سندرم کاودن ضایعات پوستی مشخص را در سن 20 سالگی نشان میدهند.
ژنتیک مولکولی :
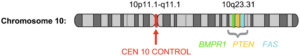
همانطور که گفته شد، جهش در ژن PTEN عامل سندرم کاودن است. PTEN ژن سرکوبگر تومور میباشد که در کنترل رشد و تقسیم سلولی ایفای نقش میکند. ژن PTEN در بازوی بلند کروموزوم شماره 10 بصورت 10q23 مستقر است.
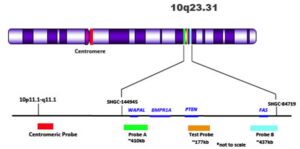
حدود 80 درصد از افراد مبتلا به سندرم کاودن دارای جهش در ژن PTEN هستند. این جهش، تنظیم حیات سلول و تقسیم سلولی را تحت تأثیر قرار میدهد که میتواند منجر به شکلگیری تومور و همچنین منجر به عدم سنتز پروتئین PTEN گردد. در 20 درصد دیگر از بیماران مبتلا به سندرم کاودن علت هنوز مشخص نیست.
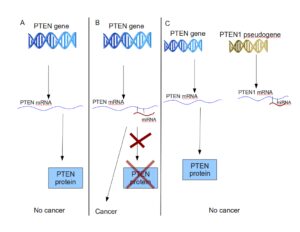
برخی از موارد ناشناخته جهش در این بیماری، ممکن است با جهش در یک منطقه از DNA که فعالیت ژن PTEN را تنظیم میکند، مرتبط باشد. دیگر افراد مبتلا به سندرم کاودن ممکن است جهش در زیرواحد خاصی از آنزیم میتوکندری بنام سوکسینات دهیدروژناز داشته باشند. به تازگی عوامل اپیژنتیک مانند فرآیند متیلاسیون در ژنهای کشنده طبیعی (NKG) همانند افراد مبتلا به سندرم کاودن، تظاهرات بالینی مشابهی را نشان دادهاند.
همهگیرشناسی :
این سندرم در حدود 1 در هر 200000 تولد زنده دیده میشود. میزان شیوع این سندرم در سراسر جهان دیده میشود. با این حال، به دلیل اینکه تظاهرات بالینی سندرم کاودن در جمعیتهای عموم جهان مشترک است، ممکن است فراوانی این سندرم بیشتر از مقدار مذکور باشد. ظهور سرطان ممکن است بسته به جنسیت بیمار متفاوت باشد، به عنوان مثال زنان مبتلا به سندرم کاودن استعداد بیشتری نسبت به ابتلای سرطان پستان و مردان مبتلا به سندرم کاودن استعداد بیشتری نسبت به ابتلای سرطان تیروئید دارند.
شروع علائم و نشانههای سندرم کاودن ممکن است در افراد مختلف، متفاوت باشد. ممکن است بعضی از افراد مبتلا به سندرم کاودن، علائم این بیماری را در زمان تولد بروز دهند و همینطور ممکن است بعضی از افراد در دهه چهارم زندگی، نشانههای بالینی سندرم کاودن را نشان دهند. در واقع سندرم کاودن یک اختلال ژنتیکی نهفته مانند سندرم ایدز میباشد و ژن جهشیافته در این سندرم مانند ویروس HIV، قادر است خود را در سلول قرنطینه کند.
علتشناسی :
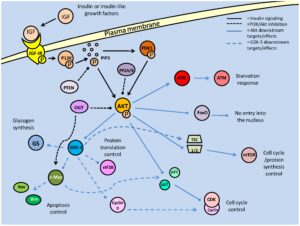
بطور کلی علت اصلی برای وقوع سندرم، جهش در ژن PTEN است که در بازوی بلند کروموزوم شماره 10 بصورت 10q23 مستقر است. اگرچه جهش در ژن PTEN دلیل اصلی بسیاری از سندرمهای کاودن به شمار میآید، اما جهشهای ژنهای دیگر نیز ممکن است باعث بروز سندرم کاودن باشند، مانند جهش در ژن BMPR1A که در بازوی بلند کروموزوم شماره 10 قرار دارد.
پاتوفیزیولوژی :
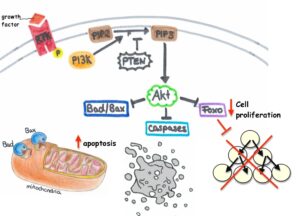
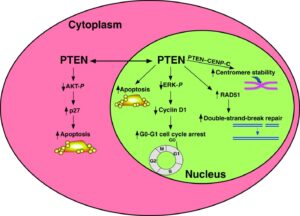
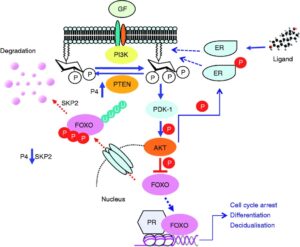
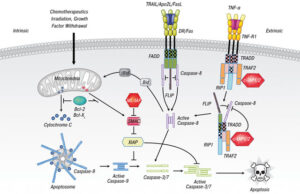
ژن PTEN ، بعنوان شناخته شدهترین ژن در عملکرد آنزیمهای فسفاتاز برای سرکوب تومورها ایفای نقش میکند. این ژن نیز بطور گسترده برای پروتئینهای TEP1 و MMAC1 ترجمه شده است. این ژن یک پروتئین بنام فسفاتاز تولید میکند که مسئول از بین بردن گروههای فسفات اضافی در سلولها و بافتهاست. علاوه بر این، ژنتیکدانان بر این باورند که ژن PTEN در مرگ فیزیولوژیک سلولی (آپوپتوزیس) نقش بسزایی ایفا میکند.
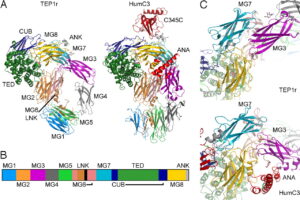
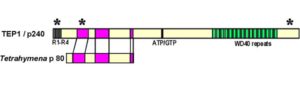
در این سندرم ، یک تغییر یا جهش در توالی ژنومی، ژن PTEN به تغییرات در فیزیولوژی ژن مرتبط است.
تاریخچه :
این سندرم برای اولین بار در سال 1963 توسط دکتر دنیس و دکتر لوید گزارش گردید.


References:
- James، William D.; Berger، Timothy G. (2006). Andrews’ Diseases of the Skin: Clinical Dermatology. Saunders Elsevier. p. 673.
- Tan، MH; Mester، J، Peterson، C، Yang، Y، Chen، JL، Rybicki، LA، Milas، K، Pederson، H، Remzi، B، Orloff، MS، Eng، C (2011-01-07).
- Kumar (2009). Robbins and Cotran Pathologic Basis of DiseaseProfessional Edition، 8th ed. Saunders، An Imprint of Elsevier.
- Schrager CA، Schneider D، Gruener AC، Tsou HC، Peacocke M (January 1998). “Clinical and pathological features of breast disease in Cowden’s syndrome: an underrecognized syndrome with an increased risk of breast cancer”. Hum. Pathol. 29 (1): 47–53.
- Kay PS، Soetikno RM، Mindelzun R، Young HS (June 1997). “Diffuse esophageal glycogenic acanthosis: an endoscopic marker of Cowden’s disease”. Am. J. Gastroenterol. 92 (6): 1038–40.
- Saslow D; Boetes C; Burke W et al. (2007). “American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography”. CA Cancer J Clin 57 (2): 75–89.
- Lloyd، KM.; Dennis، M. (1963). “Cowden’s disease. A possible new symptom complex with multiple system involvement”. Ann Intern Med 58: 136–42.
- Lloyd KM، Schammel LM. (2008). Clinical Progression of CA-MRSA Skin and Soft Tissue Infections: A New Look at an Increasingly Prevalent Disease. Arch Dermatol. 2008;144:952.
- Bhattacharjee P، Fatteh S، Lloyd KM. (2004 February). Squamous Cell Carcinoma Arising in Long-Standing Lichen Sclerosis Et Atrophicus. Journal of the American Geriatrics Society. 52(2):319-320.
- Lloyd KM، Lloyd JR. (1990 June). Palmar Pits and Multiple Trichoepitheliomas: An Association. J Am Acad Dermatol. 22:1109-1111.
- Greene RS، Lloyd KM. (1969 June). Macular Atrophy (Anetoderma of Jadassohn) Secondary to Syphilis. Cutis 5:701-706.
- Published over 190 peer reviewed journal articles book chapters 40، editor two books. CV articles 180، 177، 173، 171، and 169.
https://rarediseases.info.nih.gov/diseases/6202/cowden-syndrome
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام