سندرم هانتر
Hunter Syndrome
شاهین اسعدی (دانشجوی ژنتیک مولکولی)، دکتر علی نظیرزاده (متخصص ژنتیک)، الناز حیدری (دانشجوی کارشناسی ارشد ژنتیک)، فرح قاسمپور (محقق بیولوژی)
نگارنده مسئول: شاهین اسعدی (Geneticist)

سندرم هانتر یا موکوپلیساکاریدوز تیپ 2 یک بیماری لیزوزومی ذخیرهای ناشی از کمبود یا عدم وجود آنزیم ایدونارات-2 سولفاتاز (I2S) میباشد. لایههای ذخیره شده لیزوزوم در سندرم هانتر شامل سولفات هپاران و سولفات درماتان میباشد. این بیماری از الگوی توارثی وابسته به جنس مغلوب پیروی میکند.
علائم و نشانهها:
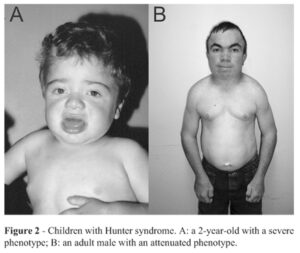
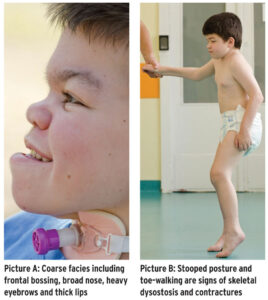
علائم و نشانههای سندرم هانتر (MPSII)، بهطورکلی در هنگام تولد آشکار است، اما معمولاً پس از پایان سال اول زندگی، بیماری مذکور مشهودتر است. اغلب، اولین نشانههای سندرم هانتر فتق شکمی، عفونت گوش، آبریزش بینی و سرماخوردگی مکرر هستند. از آنجا که این علائم در بیشتر نوزادان رایج است، پزشک معالج اغلب نمیتواند بهسادگی سندرم هانتر را تشخیص دهد، چرا که علائم سندرم هانتر مشابه عفونتهای ویروسی در نوزادان است. بیشترین نشانه سندرم هانتر، تجمع گلیکوزآمینوگلیکانها (GAG) در سراسر سلولهای بدن است. بعد از دو سالگی، نشانههای سندرم هانتر قابل رؤیت میگردد که شامل خصوصیات فیزیکی از جمله درشتی فرم صورت، پیشانی برآمده، بینی با پل پهن و زبان بزرگ است.
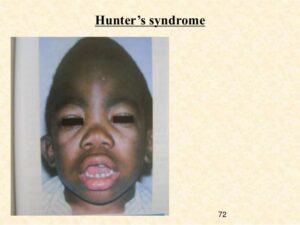
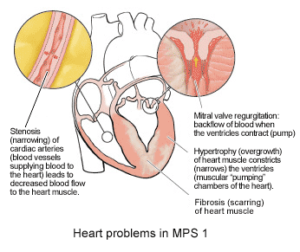
کودکان مبتلا به سندرم هانتر، دارای سر بزرگ و شکم بزرگ هستند و همچنین استعداد بیشتری برای ابتلا به عفونتهای مکرر دستگاه گوارشی، دستگاه تنفسی و گوش دارند. همه این حالتها به دلیل ذخیرهسازی بیش از حد گلیکوزآمینوگلیکانها در سلولهای بدن میباشد. در سندرم هانتر، ضخیم شدن دیواره قلب، بهتدریج میتواند در کاهش عملکرد قلب نیز نقش داشته باشد. همچنین سندرم هانتر منجر به ضخیم شدن پرده دیافراگم میشود که این حالت منجر به انسداد راه تنفسی در هنگام خواب و ایجاد مشکلات تنفسی و تنفس پرصدا و خروپف میگردد.
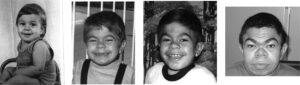
افراد مبتلا به سندرم هانتر با گذشت زمان، دارای طحال و کبد بزرگ خواهند بود، همچنین تمام مفاصل این افراد از جمله مچ دست، آرنج، شانه، باسن و زانو، بزرگ خواهد بود. سندرم هانتر منجر به سفتی مفاصل و حرکات محدود آنها نیز میشود.
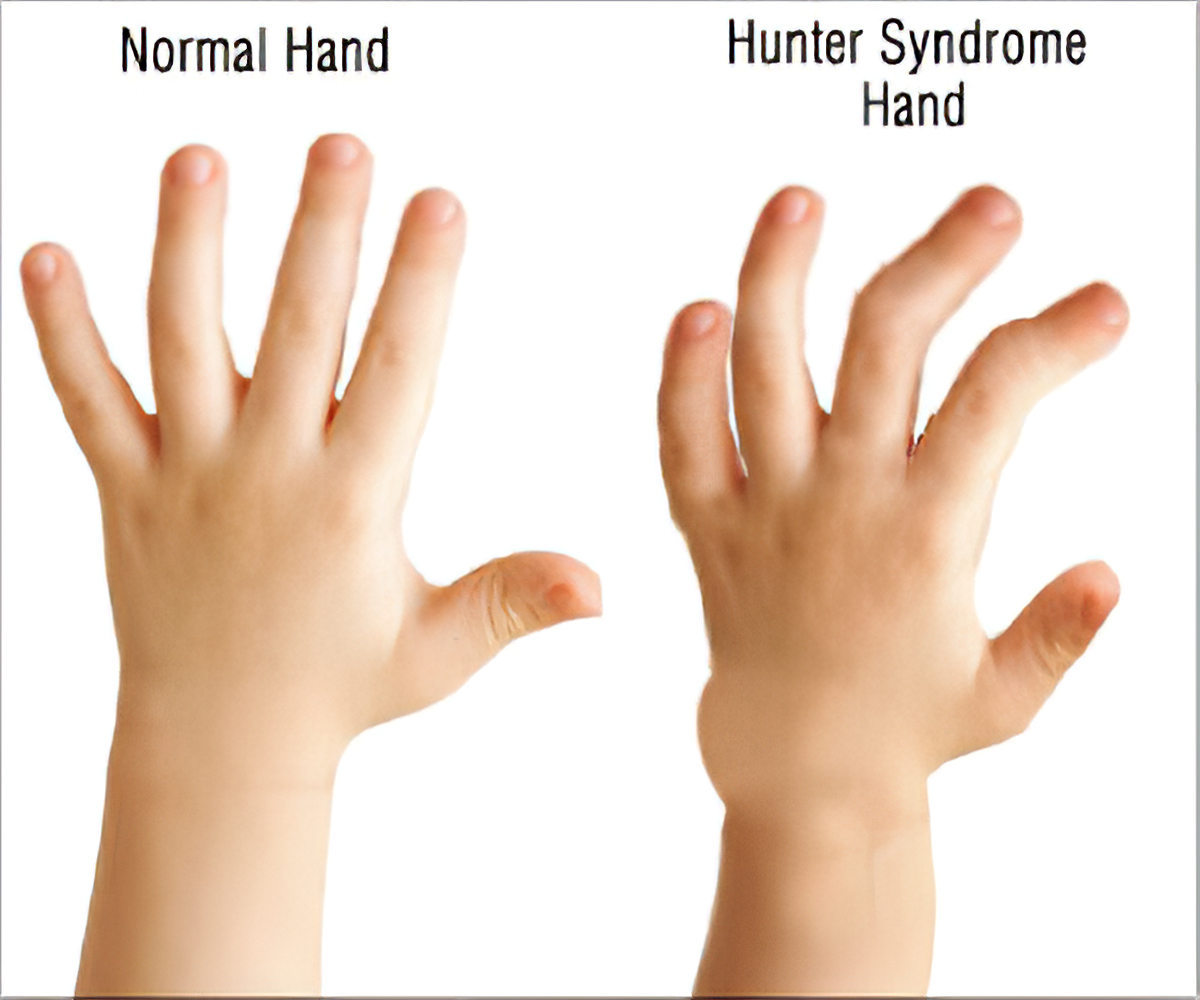
افراد مبتلا به سندرم هانتر دارای انگشت شست ضعیف جهت بلند کردن اشیای کوچک هستند. استخوانها نیز تحت تأثیر این سندرم بوده که منجر به کوتاهی قامت خواهد گردید.

علاوه بر این در افراد مبتلا به سندرم هانتر، ضایعات پوستی عاجرنگ در بازوها و ساق پاها و قسمت فوقانی پشت نیز ممکن است مشاهده شود، البته وجود یا عدم وجود این ضایعات پوستی برای شناسایی سندرم هانتر بهتنهایی مفید نیست، اما میتوان شدت بالینی سندرم هانتر را در افراد مبتلا، ارزیابی کرد. تجمع بیش از حد، GAG در مغز نیز، میتواند به کاهش رشد با عقبماندگی ذهنی بهویژه در یادگیری و آموزش در افراد مبتلا به سندرم هانتر گردد.
نرخ و درجه پیشرفت بیماری مذکور میتواند برای هر فرد مبتلا به سندرم هانتر متفاوت باشد. سندرم هانتر با طیف گستردهای از شدت بالینی به دو شکل اصلی، شدید و خفیف شناخته میشود. تفاوت بین اشکال شدید و خفیف سندرم هانتر، به توسعه تدریجی نورونها در سیستم عصبی مربوط میشود که در نوع شدید، نورونها کمتر توسعه یافته و منجر به عقبماندگی ذهنی شدید میگردد؛ اما این بدان معنا نیست که افراد مبتلا به سندرم هانتر با درجه خفیف بیماری، دارای توانایی ذهنی معتدل هستند، در واقع در نوع خفیف نیز علائم بیماری کاملاً جدی است و هرچند با درجه خفیف بیماری هستند اما باز هم دارای ناتوانی ذهنی میباشند.
با این حال برخی از افراد مبتلا به سندرم هانتر، هیچ معلولیت ذهنی ندارند و در واقع سندرم بیشتر تظاهرات فیزیکی را که شامل کوتاهی قامت و اختلال در مفاصل هست، نمایان میسازد. متوسط امید به زندگی در افراد مبتلا به سندرم هانتر از 20 سال تا 60 سال میباشد، البته این بستگی به درجه شدت بیماری دارد که هرچه شدیدتر باشد متوسط امید به زندگی، کاهش مییابد. در شدیدترین حالت سندرم هانتر، امید به زندگی میتواند از سن 15 سالگی یا حتی کمتر نیز کاهش یابد و این به دلیل اختلالات روانی حاصل از اختلالات نورونها در سلولهای عصبی میباشد. در نوع شدید سندرم هانتر، کودکان زیر 15 سال، اختلالاتی مانند بیشفعالی، اوتیسم، اختلال وسواسی جبری و یا اختلال پردازش حسی از خود بروز میدهند.

پاتوفیزیولوژی:
سندرم هانتر یک اختلال ژنتیکی است که اغلب جنس مذکر را با الگوی توارثی وابسته به X مغلوب مبتلا میکند. سندرم هانتر یکی از چندین بیماری ذخیره لیزوزومی مرتبط با موکوپلیساکاریدها و گلیکوزآمینوگلیکانها میباشد. سندرم هانتر با تجمع GAG در سرتاسر سلولها به دلیل کمبود یا عدم وجود آنزیم ایدورونات-2 سولفاتاز (I2S) همراه است. تجمع GAG در سلولها منجر به اختلالات عصبی در نورونها و سیستم اعصاب مرکزی و تأخیر در رشد میگردد. با این حال در برخی موارد، افراد مبتلا به سندرم هانتر با درجه بالینی خفیف عمر کوتاهی ممکن است داشته باشند.
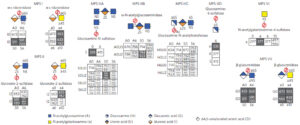
ژنتیک مولکولی
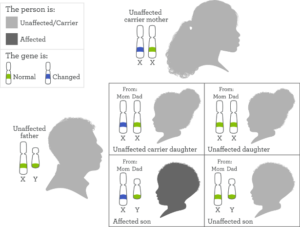
سندرم هانتر یک اختلال ژنتیکی است که از الگوی توارثی وابسته به X مغلوب تبعیت میکند و در اثر جهش ژن IDS ایجاد میشود. ژن IDS در بازوی بلند کروموزوم جنسی X بهصورت Xq27.3 مستقر است.
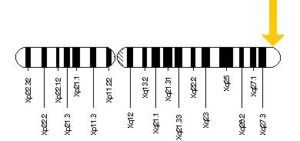
بهطور معمول هر سلول در بدن انسان، دارای 46 عدد کروموزوم است که 23 عدد از آنها را از یکی از والدین پدر و مادر به ارث میبرد. از آنجا که ژن IDS در کروموزوم جنسی X قرار دارد، بنابراین مردان مبتلا به سندرم هانتر، این ژن جهشیافته و بیماری را از مادر دریافت میکنند. مادران چون دارای دو نسخه از کروموزوم جنسی X هستند، اغلب حامل بیماری میباشند، مگر آنکه ژن IDS در هر دو نسخه کروموزوم جنسی X در خانمها دچار جهش شود؛ بنابراین زنها، در صورت داشتن دو کپی جهشیافته از ژن IDS مبتلا به سندرم هانتر خواهند بود که این حالت کمتر مشهود است.
ژنتیک بیوشیمیایی
بدن انسان به یک آرایه وسیعی از واکنشهای بیوشیمیایی و همچنین به پشتیبانی از توابع مهم مانند تولید انرژی، رشد و توسعه ارتباطات درونی با ارگانلها و ارگانها و حفاظت از عفونتهای میکروبی وابسته است. یکی دیگر از فرآیندهای مهم در بدن انسان، تجزیه بیومولکولهای درشت است که مشکل اساسی در سندرم هانتر به تجزیه نادرست این مولکولهای زیستی و اختلالات ذخیرهسازی مربوط میشود. دلیل اصلی این سندرم دقیقاً مشخص نیست اما یکی از دلایلش، افزایش سن مادر در زمان بارداری است.

بیوشیمی سندرم هانتر، به یک مشکل در بخشی از بافت همبند بهعنوان ماتریکس خارج سلولی مربوط میشود. این ماتریکس از انواع کربوهیدراتها و پروتئینها تشکیل شده که کمک میکند تا چارچوب معماری اسکلت بدن انسان، به فرم طبیعی قرار گیرد. دلیل اصلی این سندرم دقیقاً مشخص نیست اما یکی از دلایلش، افزایش سن مادر در زمان بارداری و عوامل تراتوژنیک مانند حضور اجرام میکروبی در زمان تشکیل سلول زیگوت است.
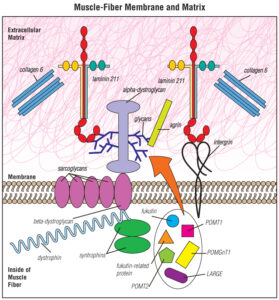
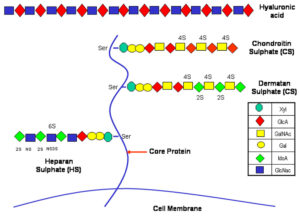
این ماتریکس تمام سلول را احاطه میکند و بهعنوان چسب، دیواره سلولها را نگه میدارد. یکی از بخشهای ماتریکس سلولی، مولکولهای پیچیدهای به نام پروتئوگلیکانها هستند. این مولکولها نیز همانند بسیاری از مولکولهای دیگر بدن، نیاز به شکسته شدن و جایگزین شدن دارند که از موکوپلی ساکاریدها ناشی میشوند؛ بنابراین با جهش ژنتیکی در ژن IDS، موکوپلی ساکاریدها بهجای سنتز گلیکوپروتئینها، گلیکوزآمینوگلیکانها را سنتز میکند که این یک خطا در فرآیند طبیعی واکنشهای بیوشیمیایی سلولهای بدن انسان محسوب میشود.
انواع مختلفی از GAG هر موجود در مکانهای مشخص بدن وجود دارد. در سندرم هانتر مشکل اساسی مربوط به تفکیک دو گروه از GAG میباشد که شامل سولفات هپاران و سولفات درماتان میباشد. برای تفکیک و تجزیه این دو گروه از گلیکوزآمینوگلیکانها، به آنزیم لیزوزومی ایدورونات-2 سولفاتاز نیاز است که در افراد مبتلا به سندرم هانتر این آنزیم یا غیرفعال است و یا اصلاً وجود ندارد. درنتیجه غیرفعال شدن یا عدم وجود این آنزیم، GAG در سراسر سلولها بهخصوص در بافتهای همبند که حاوی مقادیر زیادی از سولفات هپاران و سولفات درماتان هستند، تجمع مییابد.
تشخیص :
نشانههای فیزیکی قابل رؤیت این سندرم در افراد جوان، اولین سرنخ تشخیص اولیه میباشد. این تشخیص از زمان 2 تا 4 سال پس از تولد قابل غربالگری میباشد. پزشکان آزمونهای آزمایشگاهی بهمنظور ارزیابی فعالیت آنزیم ایدورونات سولفاتاز برای احتمال یک خطا در موکوپلی ساکارید را انجام میدهند. شایعترین آزمون غربالگری اختلال موکوپلیساکاریدوز، آزمایش ادرار میباشد. البته گاهی اوقات ممکن است آزمایش ادرار، میزان GAG را بهصورت طبیعی نشان دهد، در حالی که کودک مبتلا به یک اختلال موکوپلیساکاریدوز است. تشخیص قطعی سندرم هانتر، با اندازهگیری آنزیم I2S، در سرم، سلولهای سفید خون و یا بیوپسی از فیبروپلاست پوست قابل ردیابی است. تشخیص پیش از تولد سندرم هانتر، بهطور معمول با اندازهگیری فعالیت آنزیم I2S، در مایع آمنیوتیک و یا در پرزهای بافت جفت امکانپذیر است.
درمان تسکینی
با توجه به ماهیت بیماری هانتر و عدم درمان واقعاً کارآمد، میبایست بر اساس شدت بالینی بیماری، از روشهای تسکیندهنده بهمنظور تخفیف رنج بیماران استفاده نمود. در واقع با توجه به علائم متنوع بیماری، از عمل جراحی جهت بهبود کیفیت فرم فیزیکی اسکلتی و استراتژی رفتار خاص، به تسکین رنج این بیماران میتوان اقدام کرد.
پیوند مغز استخوان در سندرم هانتر:
کارآمدترین روش برای انتقال آنزیم I2S، به بیماران سندرم هانتر پیوند مغز استخوان برای مدت طولانی از افراد سالم میباشد. اگرچه ممکن است پیوند مغز استخوان نیز در اوایل بهبود کیفیت، حال بیماران هانتری بسیار کند باشد اما میتواند نقش مؤثری در امید به زندگی طولانیتر این افراد داشته باشد. با این حال پیوند مغز استخوان برای افراد ضعیف یا افرادی که درجه شدت بالینی شدیدی از سندرم هانتر را دارا هستند، میتواند خطرناک باشد و همچنین میتواند تهدیدکننده حیات برای این دسته از بیماران باشد. در خوشبینانهترین حالت، پیوند مغز استخوان مناسبترین روش برای بهبود کیفیت زندگی بیماران و افزایش امید به زندگی بیماران محسوب میشود.
اپیدمیولوژی
سندرم هانتر یک اختلال ژنتیکی نادر است که تخمین زده میشود در سراسر جهان حدود 2000 نفر مبتلا به این بیماری هستند. بر اساس بیوانفورماتیک سازمان بهداشت جهانی، 500 نفر از مبتلایان این سندرم در ایالات متحده آمریکا زیست میکنند. 6 نفر در ایرلند، حداقل 1 نفر در ایران، 1 نفر در عربستان سعودی، 1 نفر در شیلی، 1 نفر در پاکستان، 20 نفر در فیلیپین، 1 نفر در فلسطین، 70 نفر در کشور کره، 1 نفر در شهر کلکته هندوستان، 1 پسر در شهر شیلیگوری واقع در غرب بنگال هندوستان و 1 پسر هشت ساله در شهر گنگتوک هندوستان از سندرم هانتر رنج میبرند. بر اساس تحقیقات دانشگاه آکسفورد انگلستان، فرکانس این سندرم در حدود 1 تولد مرد به ازای هر 130000 تولد مرد میباشد. در واقع احتمالاً از هر 130000 تولد مرد زنده، ممکن است 1 مرد زنده مبتلا به سندرم هانتر باشد.
تاریخچه
سندرم هانتر اولین بار توسط، سرهنگ دوم سپاه انگلستان در جنگ جهانی اول، دکتر چالرز هانتر در سال 1917 گزارش گردید.
موارد قابلتوجه:
در 24 جولای 2004، مردی به نام اندرو وارگ 38 ساله از کشور انگلستان، پسر 10 سالهاش به نام ژاکوب را که مبتلا به سندرم هانتر بود با بالش خواب به قتل رسانید. نکته جالب اینجاست که آقای اندرو وارگ، علت مرگ پسرش را انسداد راه تنفسی به دلیل بیماری هانتر تلقی کرد. در آن زمان دادگاه جنایی انگلستان، صحت ادعای اندرو وارگ را تأئید کرد، اما یک سال پس از آن در 13 دسامبر سال 2005، ژنتیکدانان با تحقیقات خود ادعا کردند که مرگ به دلیل انسداد راه تنفسی در کودکان مبتلا به سندرم هانتر، بسیار نادر است و فقط این افراد در هنگام خواب دچار تنگی نفس بهصورت مزمن میشوند که همین امر منجر به خروپف کردن کودکان در هنگام خواب میشود.
البته ژنتیکدانان این ادعا را نیز کردند که مرگ به دلیل انسداد راه تنفسی در بیماران سندرم هانتر بیشتر در سنین بالای 30 سال این افراد اتفاق میافتد. همین موضوع باعث شد تا یک متخصص امنیت نظامی در انگلستان پرونده آقای اندرو وارگ را دوباره به دادگاه جنایی کشاند و بر اساس یافتههای ژنتیکدانان، آقای اندرو وارگ پس از یک سال از مرگ پسرش، متهم به قتل ژاکوب پسر 10 سالهاش شد. البته اندرو وارگ، این اتهام را تکذیب کرد و قاضی دادگاه خانم آنا رافرتی، اندرو وارگ را به دلیل قتل پسر 10 سالهاش که مبتلا به سندرم هانتر بود، مجرم شناخت و او را به زندان محکوم کرد.
REFERENCES:
- Wraith JE، Scarpa M، Beck M، et al. (March 2008). “Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy”. Eur. J. Pediatr. 167 (3): 267–77.
- James، William D.; Berger، Timothy G.; et al. (2006). Andrews’ Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 544.
- Muenzer، J; Wraith، JE; Beck، M; Giugliani، R; Harmatz، P; Eng، CM; Vellodi، A; Martin، R; Ramaswami، U; Gucsavas-Calikoglu، M; Vijayaraghavan، S; Wendt، S; Puga، AC; Ulbrich، B; Shinawi، M; Cleary، M; Piper، D; Conway، AM; Kimura، A (August 2006). “A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome).”. Genetics in medicine : official journal of the American College of Medical Genetics 8 (8): 465–73.
- Young ID، Harper PS (1982). “Incidence of Hunter’s syndrome”. Hum. Genet. 60 (4): 391–2.
- Hunter، C. A. (1917). “A Rare Disease in Two Brothers”. Proceedings of the Royal Society of Medicine (London) 10 (Sect Study Dis Child): 104–116.
- “A Phase I/II، Randomized، Safety and Ascending Dose Ranging Study of Intrathecal Idursulfase-IT Administered in Conjunction With Intravenous Elaprase in Pediatric Patients With Hunter Syndrome and Cognitive Impairment”. Clinicaltrials.gov. U.S. National Institutes of Health. 6/12/2009. Retrieved 2014-07-20.
- “A Safety and Dose Ranging Study of Idursulfase (Intrathecal) Administration Via an Intrathecal Drug Delivery Device in Pediatric Patients With Hunter Syndrome Who Have Central Nervous System Involvement and Are Receiving Treatment With Elaprase® – Results”. Clinicaltrials.gov. U.S. National Institutes of Health. 2013-10-31. Retrieved 2014-07-20.
- “Study of Intrathecal Idursulfase-IT Administered in Conjunction With Elaprase® in Pediatric Patients With Hunter Syndrome and Early Cognitive Impairment (AIM-IT)”. Clinicaltrials.gov. U.S. National Institutes of Health. July 2014. Retrieved 2014-07-20.
- Hopwood JJ، Bunge S، Morris CP، et al. (1994). “Molecular basis of mucopolysaccharidosis type II: mutations in the iduronate-2-sulphatase gene.”. Hum. Mutat. 2 (6): 435–42.
- Gort L، Chabás A، Coll MJ (1998). “Hunter disease in the Spanish population: molecular analysis in 31 families.”. J. Inherit. Metab. Dis. 21 (6): 655–61.
- Crotty PL، Braun SE، Anderson RA، Whitley CB (1993). “Mutation R468W of the iduronate-2-sulfatase gene in mild Hunter syndrome (mucopolysaccharidosis type II) confirmed by in vitro mutagenesis and expression.”. Hum. Mol. Genet. 1 (9): 755–7.
- Sukegawa K، Tomatsu S، Fukao T، et al. (1995). “Mucopolysaccharidosis type II (Hunter disease): identification and characterization of eight point mutations in the iduronate-2-sulfatase gene in Japanese patients.”. Hum. Mutat. 6 (2): 136–43.
تشخیص پیش از تولد سندروم داون و نقایص لولههای عصبی
سندرم اهلرز- دانلوس
برای دانلود پی دی اف بر روی لینک زیر کلیک کنید
ورود / ثبت نام